6个Meckel-Gruber综合征家系临床和分子遗传学分析
2020-09-03张蔓丽王利群李晓青陈宁潘亭亭田亚平卢彦平
张蔓丽 王利群 李晓青 陈宁 潘亭亭 田亚平 卢彦平
Meckel-Gruber综合征(Meckel-Gruber Syndrome,MKS)是一种罕见的常染色体隐性遗传的初级纤毛病,造成的胎儿畸形涉及多种重要器官,属于致死性畸形。MKS的发病率各国报道不一,由于近亲婚配的原因,以阿拉伯国家最高,约1/1 300活产婴,携带率1/18[1],欧洲报道为2.6/10万[2]。中国发病率不详,仅见一些散发病例报道。MKS在胎儿发育的早中期即可出现典型的临床表型,包括脑膜脑膨出、轴后性多指趾、双侧多囊肾等[3]。但不同MKS病例临床表型并不完全一致,甚至有些表型与其他疾病如婴儿型多囊肾(autosomal resessive polycystic kidney disease,ARPKD)等有重叠[4],容易误诊。课题组前期设计了含有113个基因的初级纤毛病panel,结合二代测序技术,对6个MKS家系查找致病基因突变位点,以此为基础提供遗传咨询,并为有生育要求的家庭进行了PGD或产前诊断,帮助其获得了无MKS疾病的下一代。
对象与方法
一、对象
1.6个MKS家系资料:收集2009年4月—2017年11月于中国人民解放军总医院妇产科就诊的临床诊断为MKS的6个家系。该6个家系夫妻双方体健,均无遗传病家族史。最后一次妊娠MKS胎儿时,妻子年龄均未超过35岁。6个家庭均于妊娠中期常规引产。家系资料见表1。

表1 家系资料Table 1 Pedigree data
夫妻双方留取外周肘静脉血,胎儿留取皮肤或肌肉组织。家系资料通过门诊及住院病历信息获得。上述样本和信息的收集均通过本院伦理委员会审查批准,并获得家属知情同意。
二、方法
1.MKS胎儿的临床诊断:MKS胎儿的临床诊断依赖超声结果[3],通常包括多囊样肾脏,并至少包括一个其他经典特征,如枕骨脑膨出、多指趾畸形或肝脏发育异常(如肝胆管板畸形)。产前诊断超声结果满足上述条件即可临床诊断MKS。
2.DNA提取:胎儿引产后立即留取直径约3~5mm组织(皮肤、肌肉)提取gDNA。留取夫妻二人外周肘静脉血各5ml。采用常规十六烷基三甲基溴化铵法(Cetyltrimethylammonium bromide,CTAB)提取血液样本gDNA,采用通用柱式法提取组织样本gDNA。严格按照试剂盒(MyGenostics,Chongqing,China)说明进行。多余组织或血液均冻存于-80℃冰箱。
3.目标外显子结合高通量测序及结果判读、验证:
(1)文库构建。上述提取的gDNA,采用MyGenostics文库构建试剂盒(MyGenostics Inc,Chongqing,China),严格按照说明书操作。首先核酸片段化,末端修复,添加A尾。第二步进行接头连接。第三步连接产物纯化。采用磁珠法纯化产物。第四步PCR扩增,随后PCR产物纯化,同样采用磁珠法纯化产物。
(2)目标区域捕获。此步采用含有113个基因的初级纤毛病panel进行检测。该panel针对纤毛病相关的113个基因的编码外显子区域及其侧翼区域开展针对性捕获富集,纯化后的DNA保存在无酶水中。使用Qubit dsDNA HS Assay Kit对文库产物进行定量,使用Agilent 2100 Bioanalyzer system(Agilent DNA 1000 Kit)测定文库片段长度。
(3)上机测序。将上述质检合格的文库于Illumina Nextseq 500测序仪上进行双端测序。
(4)生物信息学分析。原始数据下机后保存为FASTQ格式,质控后,使用cutadaptor软件(http://code.google.com/p/cutadapt/)去除测序接头、低质量序列和短序列(<80bp)。使用BWA软件(http://bio-bwa.sourceforge.net/)将质控后的序列比对到人类基因组(UCSC hg19)序列上。使用picard工具(http://broadinstitute.github.io/picard/)去除数据中的重复序列。使用GATK软件(https://software.broadinstitute.org/gatk/)的HaplotypeCaller功能获得序列中的单核苷酸多态性(single nucleotidepolymorphisms,SNPs)位点和小片段插入缺失(InDels)变异。使用GATK软件的VariantFiltration功能完成变异位点的过滤。过滤后数据以VCF形式存储并使用ANNOVA(http://annovar.openbioinformatics.org/en/latest/)软件对上述变异进行注释,重点参考1000 genome,ESP6500,dbSNP ,EXAC,Inhouse (MyGenostics),HGMD等数据库,以及PolyPhen-2,MutationTaster,SIFT,GERP++等软件的预测结果。
(5)疑似位点的Sanger验证。对于panel测出的可疑位点,合成特异性引物,Sanger测序法用ABI3730xl测序仪(美国Applied Biosystems公司)以进行测序,测序结果与目标区域捕获测序后的结果进行比对,高通量测序及一代测序验证均由北京迈基诺基因科技股份有限公司协助完成。
结 果
一、6个家系MKS胎儿表型记录
6个MKS家系,除家系1的第4名MKS胎儿和家系6的两名MKS胎儿,其余MKS胎儿均表现出经典的“三联征”-枕部脑膨出、双侧多囊肾以及轴后性多指趾(见表1)。家系1的第4名MKS胎儿合并有先天性心脏畸形和唇腭裂。图1为家系5的MKS胎儿大体解剖图,提示该名MKS胎儿除经典三联征外尚有低位耳和短颈表现。

表2 6个家系MKS胎儿临床表型Table 2 Clinical phenotypes of MKS fetuses fromsix families
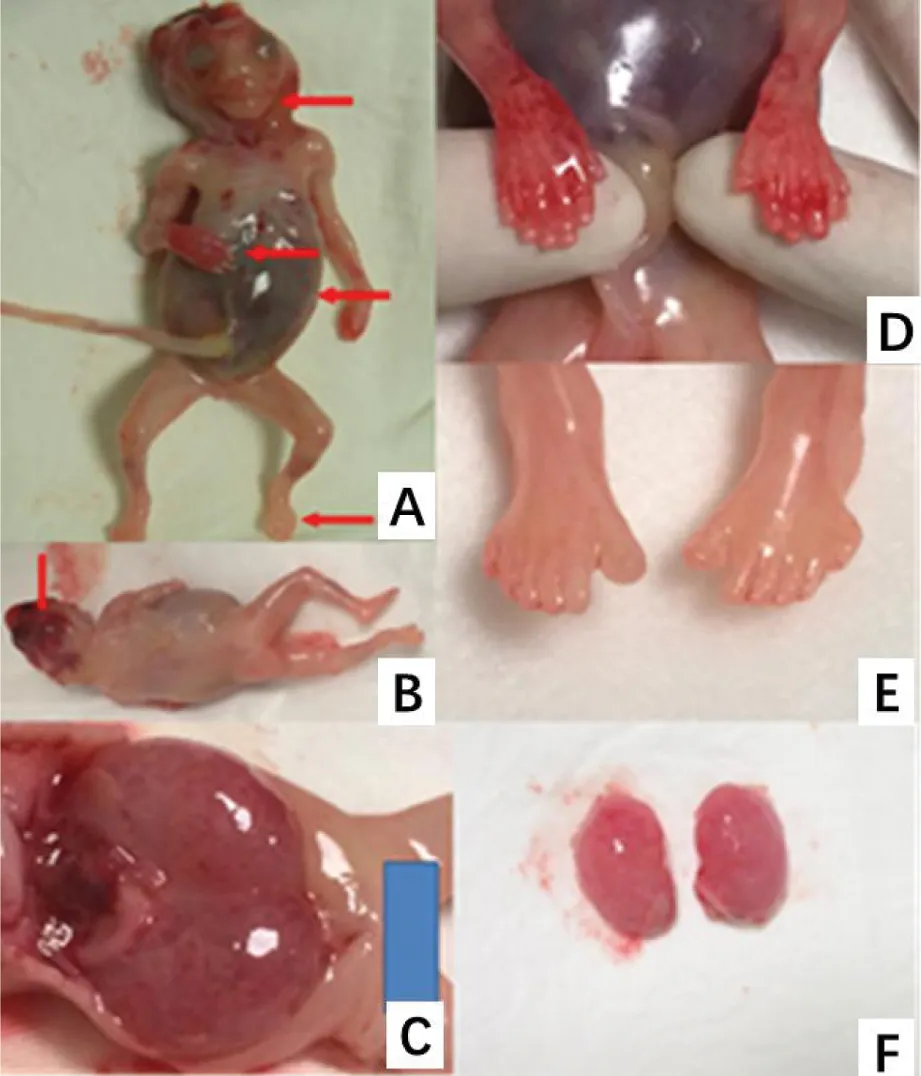
Arrows in picture A point to different malformations on MKS fetus from family 5,including low-set ear,postaxial polydactylism and enlarged abdomen.Arrows in picture B point to occipital encephalocele.Picture D and E show postaxial polydactyly.Picture C and F show enlarged abdomen was caused by bilateral polycystic kidneys.图1 家系5 MKS胎儿表型Figure 1 Clinicalphynotypes of MKS fetus from Family 5
二、二代测序及Sanger验证结果
对6个MKS胎儿进行二代测序,其中为5个家系找到致病基因及其突变(见图2)。其中家系3发现的两个突变位点CC2D2A(c.347delA,p.E116fs;c.4463_4466del,p.S1488fs)、家系4 的CC2D2A(c.1326delC,p.Q443Rfs*15)突变位点以及家系5的TCTN2(c.343G>T,p.E115X;c.1540C>T,p.Q514X)两个突变位点均未见报道,涉及突变类型为碱基缺失造成的移码突变和碱基点突变造成的无义突变。上述致病基因的两个等位基因突变均分别来源于父母,符合常染色体隐性遗传模式。
三、6个家系妊娠结局
对上述6个家系进行遗传咨询后,家系1进入PGD流程,于2012年足月剖宫产一健康男婴。家系2要求于外院行PGD,追访未有结果。家系4要求于本院行PGD,待进入流程。家系3、5要求自然受孕,家系3自然受孕后于外地建档产检,足月顺产一健康男婴。家系5成功受孕后于我院建档,孕13周行超声产前诊断提示胎儿发育正常,孕妇及家属拒绝有创检测,向其告知风险后定期常规产检,均提示正常,于2017年足月顺产一健康女婴。随访至今家系1、3、5的三名幼儿发育正常。家系6失访。

Picture A represents sequencing results for five families.Picture B,C and D respectively shows gene mutations in Family 2,3 and 4 sequenced by NGS,verified by Sanger sequencing.Due to the insufficient sample DNA from fetus,one of the gene mutations(CC2D2A c.347delA,p.E116fs) failed to be verified on fetus from Family 4 by Sanger sequencing.图2 5个家系基因测序图Figure 2 Sequencing results for five MKS families
讨 论
MKS经典三联征包括多囊肾、脑膜脑膨出、多指/趾畸形。MKS的诊断是否需要同时满足这三个表型,一直存在争议[1,5],因为不同病例报道中这三者的外显率并不一致。来自于卡塔尔的MKS流行病学调查指出此三联征在其病人中的检出率分别为84.2%,92%以及52.6%[6],而来自欧洲的人群调查报道MKS中多囊肾患病率为97.7%,脑膨出为 83.8%,多指趾87.3%[7]。本研究收集的6个MKS家系,虽然绝大多数患病胎儿表型出现了三联征,仍有一名MKS胎儿没有多指/趾畸形表型(见家系1),出现该情况的原因可能与调控基因变异相关[8]。此外家系中有些MKS胎儿还出现了小下颌、心脏畸形、腭裂等表型。因此完全依赖临床表型三联征去诊断MKS存在很大风险,必须结合基因检测进一步明确诊断。
本研究利用二代测序技术为5个家系找到致病基因突变位点。OMIM记录的MKS致病基因目前有13个,近年来TMEM237、CEP55也被报道与MKS样的疾病相关[9,10]。家系3发现的CC2D2A两个突变位点CC2D2A(c.347delA,p.E116fs;c.4463_4466del,p.S1488fs)、家系4 的CC2D2A(c.1326delC,p.Q443Rfs*15)突变位点以及家系5的TCTN2(c.343G>T,p.E115X;c.1540C>T,p.Q514X)两个突变位点均尚未报道,突变类型为碱基缺失造成的移码突变和碱基点突变造成的无义突变。CC2D2A基因位于4p15.32,编码包含1620个氨基酸的蛋白质。该蛋白1020到1205位被称为C2结构域,在物种间高度保守[11]。本研究新发现的CC2D2A三个变异位点一个位于C2结构域,两个位于N端,均涉及C2结构域改变和C端蛋白丧失。中国汉族人中该基因C2结构域和C端(1205-1620位氨基酸)单体型与智力障碍高度相关[12],侧面反映这两段结构与神经系统发育的重要关系。CC2D2A基因也是初级纤毛转化区(trasition zone,TZ)的重要蛋白,作为细胞“天线”存在的初级纤毛与肾小管发育密切相关[13]。因此CC2D2A基因移码突变影响蛋白功能,进一步导致肾小管上皮细胞初级纤毛异常,进而导致肾脏囊性样变[13]。目前对人类TCTN2基因研究和病例报道及研究极少,因此本研究对家系1[14]和家系5 MKS胎儿(数据未展示)肾脏病理切片均进行HE染色,观察肾脏结构,结果与既往国外文献报道类似,与其他基因相关的MKS啮齿类动物模型肾脏病理改变无明显区别。搜索gnomAD(http://gnomad.broadinstitute.org)数据库,发现家系5位点TCTN2 c.343G>T,p.E115X突变尚无报道,而位点c.1540C>T ,p.Q514X人群等位基因频率为4.063e-6。后续对TCTN2基因两个新突变的功能试验提示无义介导的mRNA降解(nonsense-mediated mRNA decay,NMD)机制参与了这对截短蛋白的调控(数据未展示)。TCTN2、TMEM67基因亦位于初级纤毛转化区,多篇研究指出包括CC2D2A、TCTN2、TMEM67在内的多个基因所编码蛋白在初级纤毛转化区形成NPHP 和MKS/JBTS 复合物,蛋白之间存在互作[15-16],推测这些基因的改变有可能会不同程度的影响蛋白质相互作用,从而影响共同的下游比如信号通路,造成某一类纤毛病。
通过初级纤毛疾病panel检测,未能为家系6找到致病基因。分析原因可能在于存在此次纤毛病panel未涵盖的致病基因或新基因、存在大片段缺失重复、致病位点测序质量差、生信分析过程信息被过滤、致病位点在非编码区等。2017年课题组再次使用全外显子测序技术对此家系进行分析,可惜的是仍未找到致病基因。对于此类家系,需要对测序数据每年进行数据库的再搜索,必要时使用全基因组测序。
综上所述,对MKS疾病基因的检测和致病突变的发现不仅丰富了MKS基因突变谱,为后续的遗传咨询奠定了基础,为深入研究MKS为代表的纤毛病提供了新的案例和依据。
