基于CRISPR/cas9系统高效编辑小鼠Galt基因
2020-08-28岳鹏鹏郭俊璠于鸿浩付灿王小燕高进涛
岳鹏鹏 郭俊璠 于鸿浩 付灿 王小燕 高进涛
(桂林医学院生物技术学院,桂林 541000)
近年来,CRISPR/Cas9基因编辑技术发展迅速,推动了基因工程细胞模型和动物研究的快速发展[1-4]。与应用较早的基因编辑技术如锌指蛋白核 酸 酶(Zinc finger unclease,ZFN)[5]或 转 录 激活因子样效应物核酸酶(Transcription activator-like effector nuclelease,TALENs)相比[6],CRISPR/Cas9系统具有设计灵活、成本低、操作简单、准确性高、可多位点同时打靶等优势。CRISPR/Cas9系统主要由单链向导RNA(Single guide RNA,sgRNA)和Cas9蛋白(一种核酸内切酶)组成[7]。sgRNA的主要功能是识别和结合到目标基因组DNA上,并介导Cas9蛋白切割DNA双链,最终达到基因编辑的目的。因此,sgRNA能否做到高效靶向识别和结合目标基因是CRISPR-Cas9能否特异性编辑目标基因的先决条件,尤其是基因敲除和基因敲入,sgRNA的高效性对基因打靶的影响至关重要。能够设计、制备出高效靶向目标基因的sgRNA是基于CRISPR-Cas9系统进行基因编辑的关键。
半乳糖血症(Galactosemia,GAL,OMIM #230 440)是由于半乳糖Leloir代谢途径中关键酶失活或功能异常所引起的常染色体隐性遗传代谢病,包括I型、II型和III型[8-9]。I型半乳糖血症(GAL I)在3种半乳糖血症中发病率最高[10],临床可引发一系列的症状,如肝大、肝硬化等肝损伤,随后出现白内障及肾小管功能不全等症状,严重威胁生命[11]。编码半乳糖-1磷酸-尿苷转移酶的基因GALT变异是引起GAL I的遗传病因[12-13]。目前,ARUP数据库(http://www.arup.utah.edu/database/Galt)收录了230种GALT变异类型,包括错义突变、插入突变、缺失突变、无义突变以及拼接位点突变等。GALT的变异特点具有明显的地域性,如美国和北欧地区的高频突变位点为c.563 A>G(p. Q188R)[10];东欧人群的高频位点为c. 855 G>T(p. K285N)[14];非洲人群的高频突变位点为c. 404 C>T(p. S135L)[15]。我国罕有GAL I的分子遗传学相关报道。杨茹莱等[16]报道了我国GAL I患者的GALT基因c.904 +1G>T突变为致病突变。ExAC以及ClinVar 等数据库中也记录了c. 904+1G>A和c. 904+1G>T的突变信息,且明确标注为功能失活和致病性突变。
不同GALT突变类型引起的临床症状、临床表现亦不相同。因此,有必要筛查我国明确的、典型的GALT致病突变位点,并将突变位点定位于小鼠基因组上,然后通过CRISPR/Cas9系统对该位点进行打靶,可以建立小鼠Galt基因突变细胞模型或动物模型,从而精准的模拟GAL I,为深入的理解Galt基因的致病机制以及探索可行的治疗策略具有重要意义。然而,目前并没有针对小鼠Galt基因的sgRNA导向序列以及敲除该基因的方法。鉴于此,有必要提供一种可以模拟人类致病突变的特异靶向小鼠Galt基因的sgRNA导向序列及利用其编辑小鼠Galt基因的方法,以解决现有研究的不足。
1 材料与方法
1.1 材料
1.1.1 细胞株、菌株和质粒 大肠杆菌感受态菌株DH5α和pMDTM19-T Vector Cloning Kit购 自 宝 日 医生物技术(北京)有限公司;小鼠成纤维细胞3T3、pGL3-U6质 粒 和pST1374-NLS-flag-linker-Cas9表 达质粒均由本实验室保存。
1.1.2 试剂及仪器(DNA提取试剂盒) 去内毒素质粒中提试剂盒购自北京康为世纪生物科技有限公司;LipofectamineTM 3000 Transfection Reagent(InvitrogenTM)试剂盒、杀稻瘟霉素、嘌呤霉素、胎牛血清和DMEM培养基均购自Thermo Fisher Scientific公司;Solution I、dNTP Mixture、TaKaRa Ex Taq和10×Ex Taq Buffer、Premix Taq酶均购自宝日医生物技术(北京)有限公司;BsaI购自NEB公司;DNA纯化回收试剂盒购自天根生化科技有限公司;TransDirect® Mouse Genotyping Kit购自北京全式金生物技术有限公司;引物由上海百力格生物技术有限公司合成,测序由广州生工生物工程股份有限公司完成;实验所需的主要仪器包括CO2培养箱、台式离心机、PCR仪、低温冷冻离心机、摇床等。
1.2 方法
1.2.1 sgRNA设计及sgRNA表达载体的构建 GAL I是由GALT基因突变引起的,首先利用ExAC数据库筛查了人GALT基因的功能失活突变,共筛查到19个突变位点(表1),利用OMIM在线数据库检索到GALT突变17种(表2);然后,利用ClinVar数据库检索拼接位点突变的致病情况(表3)。鉴于人GALT基因突变具有明显的地域特性,查找相关文献,找到在我国出现的、明确的、典型的致病突变(表4);最终确定了人GALTc.904 +1G为拟突变位点。
由于人和小鼠的物种差异,同一功能基因的基因结构可能不同,因此本研究利用Ensembl数据库查询了人GALT和小鼠Galt基因的结构,发现人GALTc.904 +1G位点与小鼠Galt基因c.847 +1G位点相对应(图1)。然后从Ensembl数据库中导出小鼠Galt基因序列,利用Vector NTI软件定位c.847 +1G位点序列,然后在其附近设计基因打靶位点(图1),筛选出3条sgRNA导向序列。

表1 ExAC数据库中人GALT基因功能失活突变情况

表2 OMIM数据库中人GALT基因突变情况
在上述的特异靶向小鼠Galt基因的sgRNA导向序列的5'端加上accg合成得到正向寡核苷酸序列。同时合成其对应的DNA互补链,并且在DNA互补链的5'端加上aaac合成得到反向寡核苷酸序列。将正反向寡核苷酸序列退火,形成具有粘性末端的双链DNA片段。退火反应体系为:10 μmol/L正向寡核苷酸5 μL;10 μmol/L反向寡核苷酸5 μL;10×T7 Endonuclease I buffer 2 μL;ddH2O 8 μL;反应程序为:95℃,5 min;95℃到85℃,-1℃/循环,共10个循环;85℃到25℃,-0.1℃/循环,共600个循环,退火产物-20℃保存。
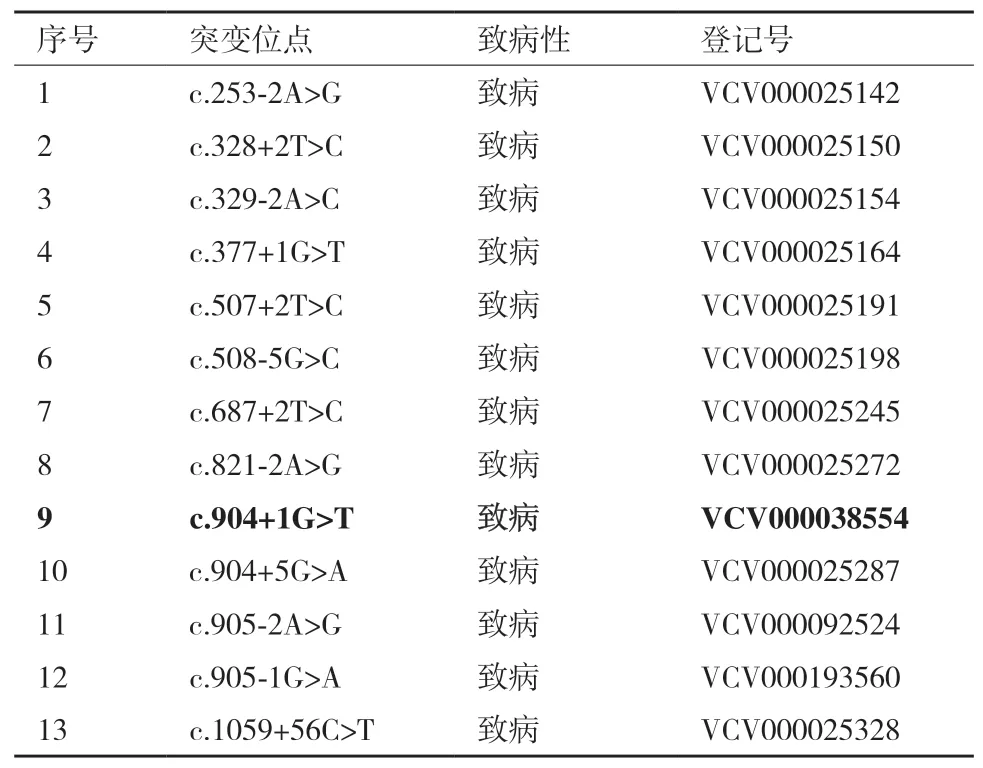
表3 ClinVar数据库中人GALT基因拼接位点致病突变情况

表4 中国I型半乳糖血症患者GALT致病突变情况

图1 模拟人GALT基因突变的小鼠Galt基因打靶位点示意图及其序列
利用BsaI限制性内切酶酶切载体pGL3-U6-sgRNA,得到酶切产物pGL3-U6-sgRNA-Bsa I,将其和上述具有粘性末端的双链DNA片段连接,并将连接产物转化感受态大肠杆菌(DH5α)并涂布于氨苄抗性的LB培养基上,37℃过夜培养20 h后,挑取单克隆并用通用引物U6测序,鉴定出阳性克隆,于37℃、220 r/min对阳性克隆摇菌,去内毒素中提试剂盒提取质粒,得到pGL3-U6-Galt-sgRNA质粒。
1.2.2 细胞转染及筛选 小鼠3T3细胞接种培养于含5%(V/V)胎牛血清的DMEM完全培养基中,于37℃,5% CO2培养箱中培养,每2-3 d更换新鲜培养基,待细胞汇合度达到80%-90%后,以0.25%胰蛋白酶消化并传代,然后分至6孔板中,16 h-18 h后,待细胞汇合度达到80%-90%时进行转染。
采用LipofectamineTM3000 Transfection Reagent(InvitrogenTM)试剂盒共转染pGL3-U6-Galt-sgRNA质粒和pST1374-NLS-flag-linker-Cas9表达质粒。每个孔(直径34.8 mm)转染以上两种质粒各2.5 μg。转染6 h后用PBS清洗,换完全培养基。培养24 h后换培养基,同时加入5 μg/mL的嘌呤霉素和10 μg/mL的杀稻瘟菌素进行药物筛选,得到阳性的sgRNA-Cas9共转染细胞。
1.2.3 基因组DNA提取及打靶位点DNA的PCR扩增 将药筛得到的阳性的sgRNA-Cas9共转染细胞进行裂解释放基因组DNA,以细胞裂解液为模板进行打靶位点DNA的PCR扩增反应。PCR扩增反应的体系为:细胞裂解液2 μL,上游引物1 μL,下 游 引 物1 μL,dNTP Mixture 2 μL,TaKaRa Ex Taq 1 μL,10×Ex Taq Buffer 2.5 μL,灭菌水补充至25 μL;PCR扩增反应的程序为:95℃,5 min;95℃,20 s,57℃,20 s,72℃,25 s,共35个循环;72℃,5 min,16℃,∞。其中上游引物序列为5'-gtgatggtgagtctcctgggta-3',下游引物序列为5'-cctttgtcagccttcagtctag-3'。
1.2.4 测序及TA克隆 取打靶位点DNA的PCR扩增产物进行Sanger测序,如果打靶位点出现套峰,则初步确认发生了基因编辑。
通过TA克隆测序分析经初步确认发生了基因编辑的打靶位点的基因型,并计算编辑效率。将打靶位点的PCR扩增产物进行胶回收纯化,将纯化后的DNA与pMDTM19-T 进行连接,连接反应的体系为:PCR纯化产物40 ng、Solution I 2.5 μL和pMDTM19-T载体0.5 μL,加水补充至5 μL。然后16℃金属浴1 h,得到连接产物。将连接产物转化感受态大肠杆菌并涂布于氨苄抗性的LB固体培养基上,37℃过夜培养20 h后,进行菌落PCR反应验证,筛选出阳性克隆,并将阳性克隆进行Sanger测序。
2 结果
2.1 sgRNA表达载体成功构建
本研究设计的sgRNA在Galt基因上的靶序列符合5'-N(21)GG的序列排列规则,且在Galt基因上的靶序列是唯一的。筛选出的3条sgRNA的核苷酸序列分别为:sgRNA1:5'-agccttaccatgccagccca-3';sgRNA2:5'-ctccatgggctggcatggta-3';sgRNA3:5'-ggctggcatggtaaggcttt-3'。按本研究实验方法构建的pGL3-U6-Galt-sgRNA质粒电泳图(图2),3种质粒大小符合预期。质粒的测序峰图(图3),阴影部分代表质粒中sgRNA编码序列,与设计的3条sgRNA相符,表明sgRNA表达载体初步构建成功。

图2 sgRNA表达质粒电泳图

图3 sgRNA表达质粒测序峰图
2.2 阳性细胞的获得及打靶位点DNA的PCR扩增
转染后,利用嘌呤霉素和杀稻瘟菌素组合可以筛选出sgRNA-Cas9的阳性转染细胞。图4为加药48 h后sgRNA1-Cas9、sgRNA2-Cas9和sgRNA3-Cas9转染细胞图。将筛选出的细胞用PBS清洗,加入胰蛋白酶消化并离心收集,裂解细胞进行打靶位点DNA的PCR扩增。打靶位点DNA的PCR扩增电泳图如图5所示,扩增条带清晰,片段大小符合预期,扩增效果效率较高,可用于后续测序分析。

图4 sgRNA1-Cas9、sgRNA2-Cas9、sgRNA3-Cas9转染阳性细胞

图5 打靶位点DNA的PCR扩增电泳图
2.3 测序及打靶效率分析

图6 sgRNA打靶位点DNA的PCR产物测序峰图
sgRNA打靶位点DNA的PCR产物测序峰图见图6。3种sgRNA靶点序列均出现套峰,且无原序列,初步表明发生了基因编辑,且编辑效率较高。通过TA克隆可将单条PCR产物连接到载体上,再通过测序分析可获得打靶位点的基因型。本研究sgRNA1、sgRNA2和sgRNA3三条sgRNA打靶位点的基因型分析见表5、6、7。3种sgRNA导向序列均可以介导Cas9蛋白,高效、特异性的切割靶点DNA,进而编辑小鼠Galt基因,效率高达100%,编辑后基因型以缺失突变为主。

表5 sgRNA1靶点基因型分析
3 讨论
一个基因的突变有很多种,包括碱基的置换、移码突变、缺失和插入突变等,且均有可能是致病突变。但在临床中表现有可能不同,即疾病的临床症状与基因的突变位置及突变类型均有关系,其机理在很多疾病中均未阐明。这也是文章中首先进行检索和筛查,确定拟突变位点的原因所在。本研究首先筛查ExAC数据库确定了GALT基因功能失活突变可导致GAL I,又通过OMIM在线数据库检索到GALT的突变种类,再通过ClinVar数据库确认具体哪些点突变会致病。但致病突变具有地域性,因此本研究继续查找相关文献,经过层层比对确定了可模拟我国GAl I致病突变的人GALTc.904 +1G为拟突变位点。为了将此位点对应于小鼠Galt基因,又查询比对了人GALT和小鼠Galt基因的结构发现,小鼠Galt基因c.847 +1G位点与拟突变位点相同,最终创新性设计了精确模拟我国人GAl I致病突变位点的靶向小鼠Galt基因的sgRNA序列。本研究确定的拟突变位点为GALTc.904 +1G,突变类型为碱基置换(G>A),由于该位点为基因的拼接位点,在外显子拼接和内含子去除的过程中发挥作用,碱基置换突变后导致外显子无法拼接过程发生异常,进而影响蛋白功能,造成疾病的发生。确实本研究的结果表明大部分突变为缺失突变,突变类型与数据库中的报道不同,但突变后同样可以造成外GALTc.904 +1G位点外现在拼接异常的效应,因此推测可以模拟数据库中报道的基因突变效应。此外,我们下一步制备基因编辑动物时,会在本研究的基础上设计碱基置换的基因编辑方案,如使用碱基编辑器系统或设计重组同源臂等,力图使得基因编辑的结果与数据库报道一致。

表6 sgRNA2靶点基因型分析

表7 sgRNA3靶点基因型分析
本研究建立的CRISPR/cas9系统实现了100%的高编辑效率。除了sgRNA设计,试验中还注意了转染前细胞状态、转染试剂盒、药筛浓度和细胞DNA提取试剂盒等试验细节的优化。例如,预试验确定转染前细胞汇合度在70%-80%时的转染效率较高;转染试剂盒使用的LipofectamineTM3000 Transfection Reagent(InvitrogenTM),其转染效率高于实验室前期使用的LipofectamineTM2000;药筛浓度的控制直接决定阳性细胞获得率,通过预试验优化了最佳药筛浓度。药筛后阳性细胞量较少,实验中未使用普通基因组DNA提取纯化试剂盒,改用全式金TransDirect® Mouse Genotyping Kit,其独特裂解液裂解细胞后无需纯化,裂解物可直接作为模板进行后续扩增,提高了实验效率。多项细节优化获得了100%的编辑效率。
本研究设计的sgRNA导向序列可以介导Cas9蛋白,高效、特异性地切割靶点DNA,用于编辑小鼠Galt基因,进而影响小鼠Galt基因编码蛋白的功能。利用特异靶向小鼠Galt基因的sgRNA导向序列,构建了可模拟人类GALT基因的致病突变的CRISPR/Cas9系统,实现了小鼠3T3细胞的高效转染,确定了合适的阳性细胞药物筛选浓度,并实现了微量细胞的基因型分析。3T3细胞是常用的一种小鼠细胞模型。由于小鼠是实验动物,且多为近交系品种,因此3T3细胞的基因组DNA序列与我们将要使用的研究对象C57/B6J小鼠差异很小,且通过数据库比对分析发现3T3细胞和C57/B6J小鼠的Galt基因打靶区域序列并无差异。因此,可以认为设计的sgRNA序列在制备基因编辑动物时发挥同样的基因编辑效力。总之,本研究为建立精准模拟I型半乳糖血症的动物模型奠定了基础。
GAL I是一种影响肝脏、肾脏、生殖系统及神经系统等多器官、多系统的,具有复杂病理过程的隐性遗传代谢疾病,严重影响患者健康,甚至威胁生命[17]。目前,GAL I的发病机制仍未完全阐明[18],治疗手段单一,尚无良好的办法治疗GAL I的慢性并发症[19-20]。在本研究构建的CRISPR/Cas9系统基础上,项目组下一步将通过显微注射受精卵的方式,构建精准模拟GAI I的小鼠模型,用于非医疗诊断或治疗目的,对研究Galt功能、GAL I致病机理以及相关生物治疗方法等具有极其重要的作用。
4 结论
本研究首先分析了我国I型半乳糖血症GALT基因的致病突变位点,并将其定位在小鼠Glat基因上,然后根据小鼠Galt基因拟突变位点区域的序列设计了sgRNA导向序列,建立了高效编辑小鼠Galt基因的CRISPR/Cas9系统,编辑效率为100%。
