神经酰胺从头合成途径关键代谢酶的研究进展
2020-08-19李玉慧
李玉慧, 陈 成
(天津大学 生命科学学院, 天津 300072)
神经酰胺(Ceramide)由神经鞘氨醇长链碱基和可变长链的脂肪酸组成[1-2](图1),神经鞘氨醇长链碱基和不同的脂肪酸组合形成不同的神经酰胺,因此神经酰胺不是一种物质,而是结构相似的一类物质,脂肪酸链的长度、不饱和数以及位置都会影响神经酰胺的性质。神经酰胺不仅是真核生物细胞膜的重要组成成分,还在细胞信号传递过程中充当第二信使的重要角色[3],在细胞增殖、细胞凋亡、细胞分化等细胞代谢中发挥着十分重要的生物学作用。正是因为神经酰胺参与了众多体内代谢活动,所以说其含量一旦异常,便会对个体健康造成极其严重的影响[4-5]。
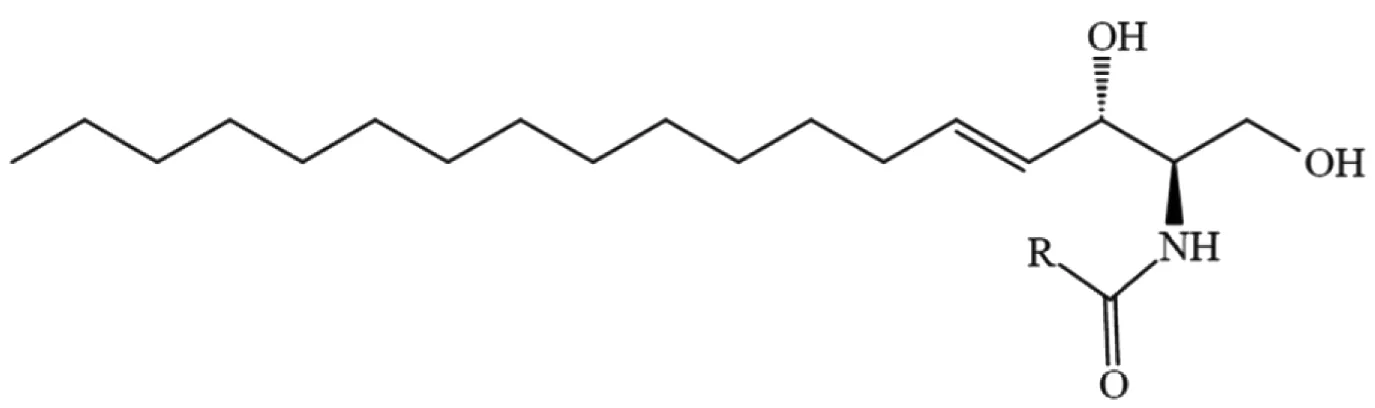
图1 神经酰胺化学结构式
神经酰胺的合成主要包括3条途径,分别为:从头合成途径、鞘磷脂酶途径以及补救途径[4-5](图2)。从头合成途径以丝氨酸棕榈酰转移酶(Serine palmitoyltransferase,SPT)将L-丝氨酸和棕榈酰-CoA缩合为3-酮二氢鞘氨醇为起始步骤,之后3-酮二氢鞘氨醇还原酶(3-Ketodihydrosphingosine reductase,KDSR)将3-酮二氢鞘氨醇还原为二氢鞘氨醇,随后二氢鞘氨醇在神经酰胺合酶(Ceramide synthase,CERS)的作用下生成二氢神经酰胺,最后二氢神经酰胺被二氢神经酰胺去饱和酶(Dihydroceramide desaturase,DES)作用生成神经酰胺;鞘磷脂酶途径由鞘磷脂酶(Sphingomyelinase,SMase)将鞘磷脂水解生成神经酰胺;补救途径先将复杂的鞘脂转化为鞘氨醇,然后通过鞘氨醇的再酰化生成神经酰胺。
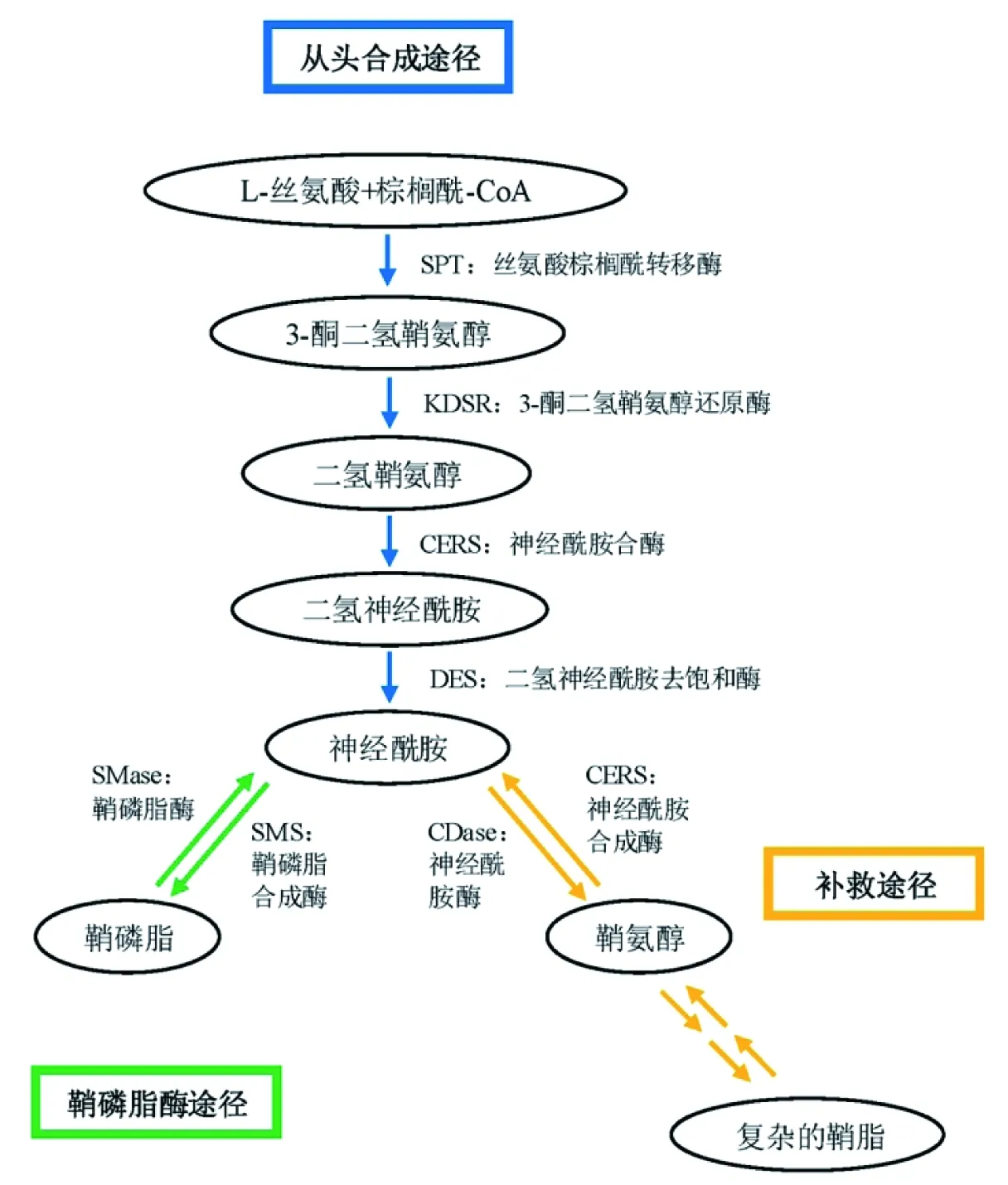
图2 神经酰胺的合成途径示意图[6]
从图2中可以看出,神经酰胺的从头合成途径可以为调控其水平提供更多可能的靶点,因此目前对该途径中的关键代谢酶的研究越来越多。2019年5月和7月分别发表在Cell和Science上的两篇文章,就CerS6和DES1进行深入研究后表明了它们与肥胖症和糖尿病的重大关联,说明了神经酰胺从头合成途径中代谢酶的重要作用,由此本文主要对该合成途径中涉及的4种酶的结构,功能,疾病相关性的研究进展进行综述,旨在为深入研究该条途径提供理论参考。
1 SPT的研究进展
SPT属于α-氧胺合成酶(AOS)家族,该家族是磷酸吡多醛(PLP)依赖性的酶。SPT负责将L-丝氨酸和棕榈酰-CoA脱羧缩合生成3-酮二氢鞘氨醇[7](图3),这一步是限速步骤,在所有的生物体内保守。

图3 丝氨酸棕榈酰转移酶催化的反应[7]
真核生物中,SPT是定位在内质网上的酶,是由SPT1和SPT2构成的异二聚体,SPT2包含PLP结合所需的赖氨酸残基,SPT1缺少赖氨酸残基[8-9],但是这两个亚基对于产生功能活跃的异二聚体均是必需的。尽管SPT1在异二聚体中具有明显的非催化作用,但已显示该亚基高度保守区域内的单个氨基酸突变会下调SPT活性,从而导致遗传性感觉神经病1型疾病(HSAN1,ORPHA:36386)[10-11]。对HSAN1患者分析显示,SPT1的两个突变热点分别为Cys133(C133W或C133Y)和Val144(V144D)[12]。
研究人员报道了鞘氨醇单胞菌中结合PLP的SPT高分辨率结构[12], SPT单体由3个结构域组成:N端结构域、催化中心和C端结构域(图4),同时研究还表明PLP与Lys265结合,也就是说Lys265对发挥酶活至关重要[12]。因为该研究是SPT结构的首次报道,不仅为分析突变位点的致病机制提供了结构基础,还为之后的研究提供了方法学上的指导[13],为更好地研究该家族的催化机制提供了见解。该研究组使用已解析的结构对人类酶建模,并将人类SPT1的Cys133映射到细菌SPT的Asn100上,将Val144映射到ASP111上,发现二者均靠近二聚体界面(图4-A),因此突变会导致复杂的结构变化,进而影响酶活性[13]。SPT除了可以利用L-丝氨酸作为底物外还可以利用其它的氨基酸,研究表明突变使SPT失去了对丝氨酸的底物偏好性,导致丝氨酸和非丝氨酸衍生的鞘脂不平衡,所以出现了疾病HSAN1[14]。这两项研究不得不使我们产生思考:突变可以使结构产生变化,同时突变也导致了SPT底物偏好性的变化,那么SPT的底物特异性和结构之间究竟是如何关联的?
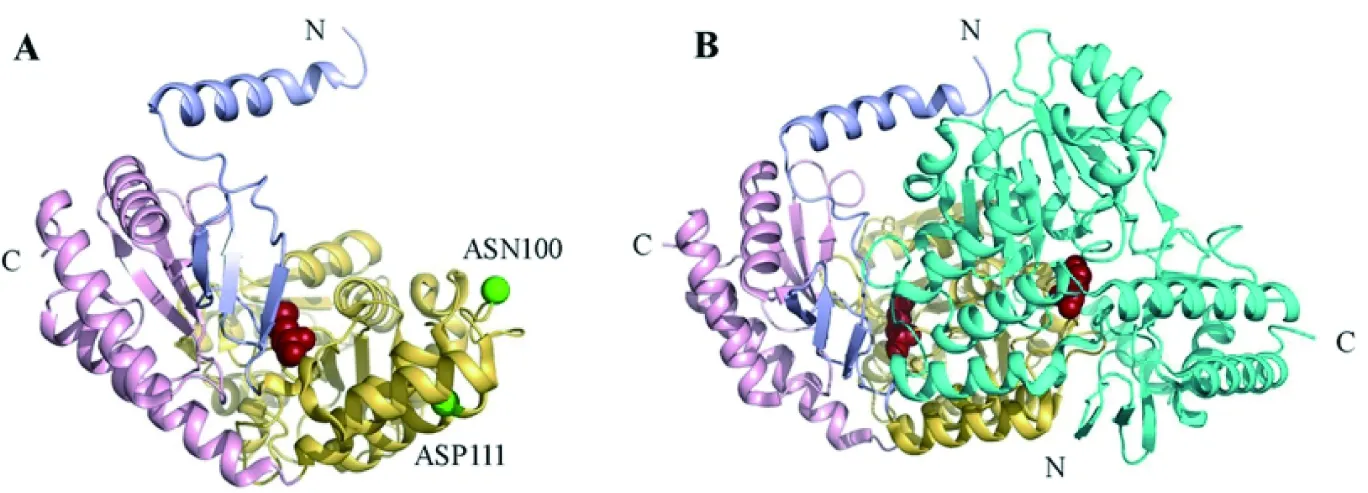
A为SPT的单体结构图。N端用浅蓝色表示,C端用浅粉红色表示,中间包含催化中心的区域用黄橙色表示,PLP的结合位点用砖红色小球表示;映射到细菌上的突变位点ASN100和ASP111用绿色球体表示。B为SPT异二聚体的整体结构图。一个单体的颜色分布和图A相同,另一个单体用青色表示。N端和C端分别用“N”和“C”标记
虽然目前已经解析了细菌中SPT的三维结构,也确定突变会导致SPT的底物偏好性产生变化,但是突变影响SPT底物偏好性产生变化后的下游事件尚不清楚,即信号是如何被逐级放大的还需要进行更深入的研究。
2 KDSR的研究进展
KDSR在神经酰胺从头合成途径中的主要功能是在还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)的协助下将3-酮二氢鞘氨醇还原为二氢鞘氨醇(图5)。
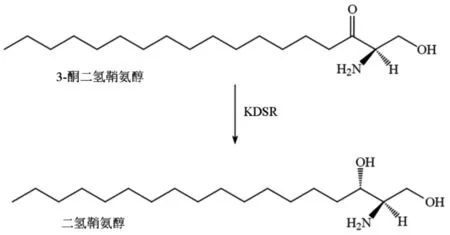
图5 3-酮二氢鞘氨醇还原酶催化的反应
KDSR属于短链脱氢酶,是位于内质网上的3次跨膜蛋白,N端具有一个跨膜区,即1~21位氨基酸。C端包含两个跨膜区,分别为271~291位氨基酸和294~314位氨基酸,中间部分(22~270位氨基酸)是包含酶活性位点(186~190位氨基酸,YXXXK)的片段,面向内质网外的胞质侧[15](图6-A)。目前尚无KDSR的三维结构信息,但是我们利用结构预测网站对其进行了预测,结构中α螺旋和β折叠交替排列形成典型的罗斯曼折叠:β-α-β-α-β,符合脱氢酶家族的结构特点(图6-B)。
研究人员发现了进行性对称性红斑角化症(Progressive symmetric erythrokeratoderma,PSEK,ORPHA:316)的致病因子为KDSR[6],这一发现暗示了神经酰胺类信号分子的功能失常和角化症的潜在联系[16]。 另一项研究[17]表明KDSR 病理突变除可导致皮肤角化症外,还可导致血小板减少症,进一步表明了 KDSR引发PSEK病理机制的复杂性。Bariana等[18]发现了KDSR的另一突变位点R154W,并利用斑马鱼模型确认了KDSR突变会导致血小板减少,Park等[19]同样有利用斑马鱼模型进行研究,发现了KDSR突变I105R诱导斑马鱼脂肪变性并产生肝损伤。近几年研究中涉及的部分突变如表1所示,但是目前并没有明确的基因型与表型相关性的研究,同时可以发现,近年来关于KDSR的研究所用的模式生物均为斑马鱼,但其实在此类研究中更偏向的模式生物为小鼠,这也为我们的研究提供了一个思路。
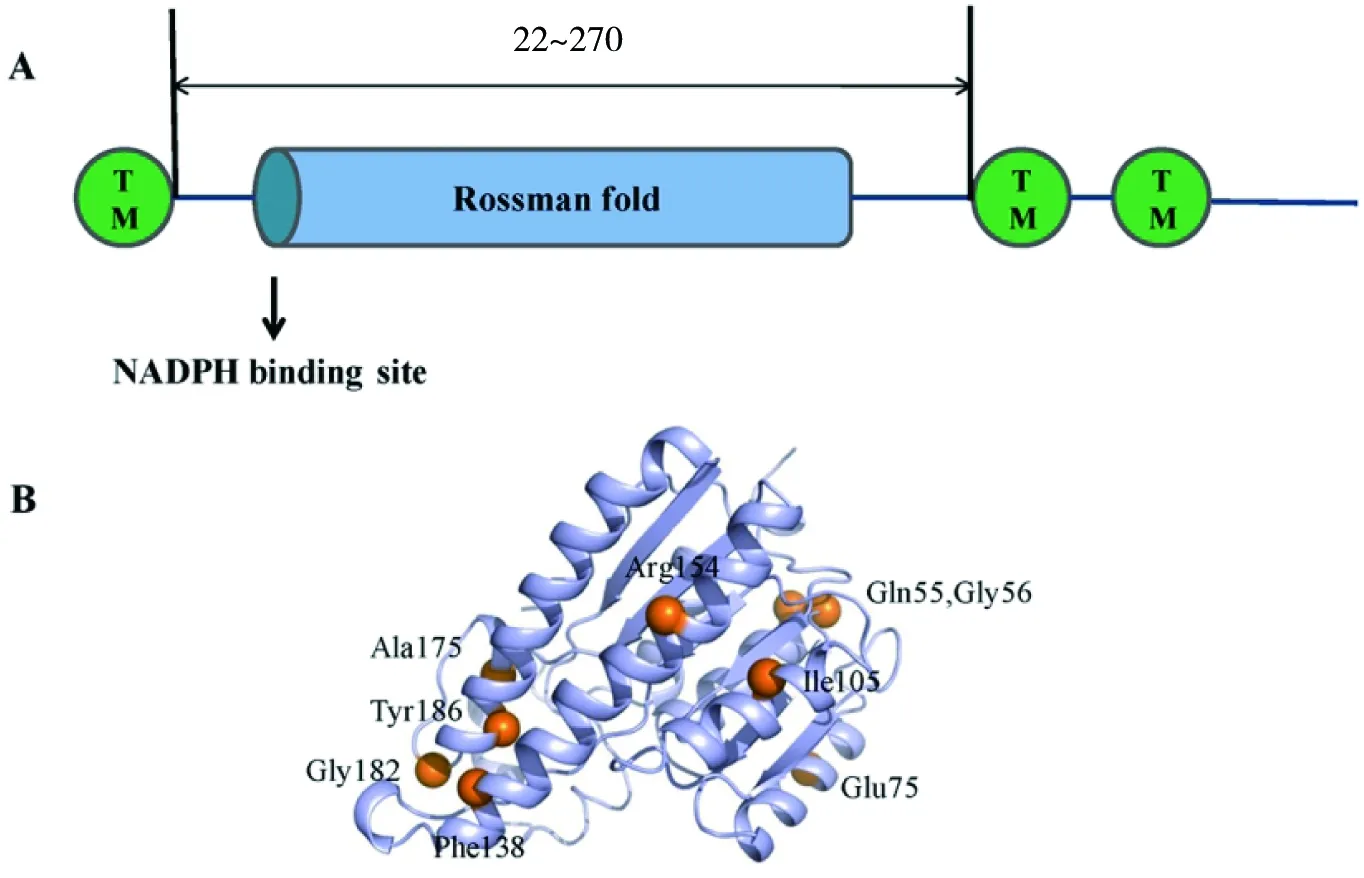
A为KDSR的拓扑结构图[6];B为KDSR的三维结构预测图。部分突变位点用橙色球体表示
KDSR是本实验室在神经酰胺从头合成途径中研究的酶,对其的研究主要集中在结构和功能上,目前其结构尚在优化之中,但是可以看出和之前预测的结构基本保持一致。我们尝试利用小鼠进行KDSR的功能性研究,在小鼠体内将编码KDSR的基因敲除,本意是想确定KDSR缺失后对下游信号通路产生的影响,但是敲除后直接致使小鼠死亡,虽然导致后续实验无法进行,但这也正说明了哺乳动物体内KDSR的重要性。
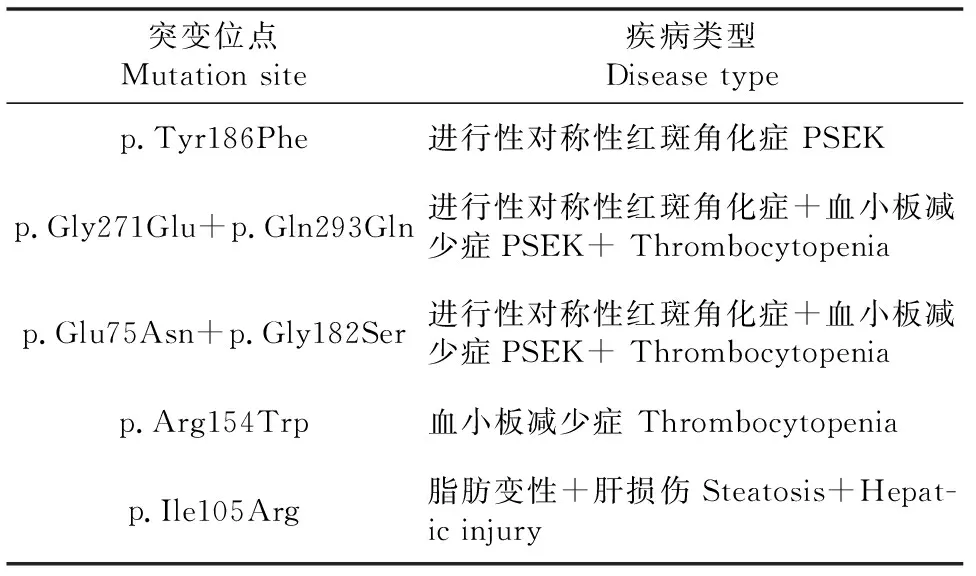
表1 KDSR中的突变位点及其导致的疾病类型
3 CERS的研究进展
CERS负责将二氢鞘氨醇N-酰化以形成二氢神经酰胺(图7)。哺乳动物中的CERS有6种,均定位在内质网中[20],每种都使用特定链长的酰基-CoA生成二氢神经酰胺[18](表2)。
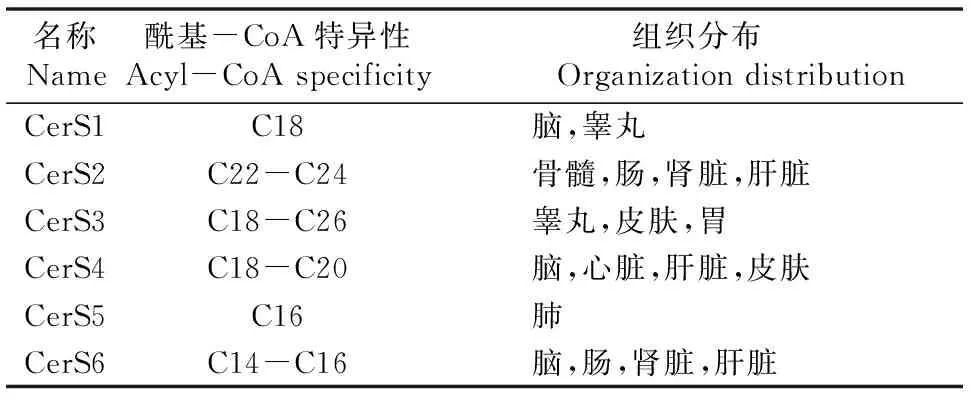
表2神经酰胺合酶的酰基-CoA特异性以及组织分布[21]

图7 神经酰胺合酶催化的反应
研究表明,CERS具有6个跨膜区(TMD)[22],第4个TMD不能完全跨过内质网膜(图8)。N端和C端位于相对两侧[20, 23],N端面向内质网腔,包含糖基化位点。C端面向胞质侧并含有磷酸化位点[24-25],CERS的磷酸化可能是控制各种酰基链长度的鞘脂的分布和水平的关键调控点,中间的结构域包括Hox样结构域、TLC结构域、Lag1p基序。6种CERS为同工酶,但是可以明显看到CerS1缺少Hox样结构域,那么该结构域的重要性、发挥作用的机制尚需深入探索。
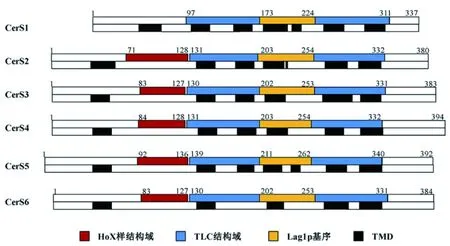
图8 CERS的域结构图[21]
哺乳动物体内的6种神经酰胺合酶具有底物特异性,组织分布也不尽相同(表2),并且其含量异常时也会对个体产生影响。CerS1基因异常会导致8型进行性肌阵挛性癫痫(ORPHA:424027);CerS2是一种抑癌蛋白,缺乏CerS2的小鼠特征是所有组织的C24:0和C24:1神经酰胺都被消耗掉,并在7~10个月大的时候发展为肝腺瘤(HCA)和肝细胞癌(HCC)[26-27];CerS3的关键生理功能已被定位在睾丸和皮肤中[28],CerS3完全缺乏的小鼠在出生后不久便会死于皮肤病[29],CerS3基因突变的人易患先天性非球形鱼鳞状红皮病(ORPHA:79394 )以及鱼鳞病-矮小身材-短毛-微球菌综合征(ORPHA:363992);人乳腺癌和肝癌[30]的恶性转化和生长伴随着CerS4表达的升高;CerS5被确定为结直肠癌(CRC)的标志物[31]。除了癌症之外,2016年的一项研究通过遗传抑制CerS5可以减少内源性C16:0神经酰胺以缓解肥胖症及其并发症,表明了CerS5和肥胖症之间的关系[32]。在胃癌中,CerS6被描述为一种新型癌蛋白,之后进一步证明了无论在体外还是体内,CerS6基因敲除均可减少胃癌细胞的增殖、迁移和侵袭[33]。关于CerS6的最新研究进展是发表在Cell上的一项研究[34],小鼠在高脂肪饮食中变肥胖时,使CerS6在小鼠体内的基因失活能对肥胖、脂肪肝和胰岛素抵抗产生保护作用,进一步研究发现与CerS6产生的C16:0神经酰胺相互作用的线粒体分裂因子Mff对于CerS6的调控作用,为肥胖症的治疗提供了新方向。值得一提的是CerS2是一种抑癌蛋白,而其它几种CERS均更倾向导致疾病的发生,如果利用组织分布的不同进行解释,其它几种CERS虽然都致病但是组织分布不同,好像是自相矛盾的;如果以域结构为出发点进行解释,CerS3、CerS4、CerS5、CerS6的域结构排布相似,也不具有说服力。那么究竟是什么原因导致了CerS2与其它几种CERS的功能相反也是今后需要致力研究的一个方向。
由于CERS为多跨膜蛋白,将其从内质网中提取出来进行纯化结晶非常困难,所以目前无可用的三维结构,但是如果想要深入了解CERS与疾病之间复杂的调控机制就必须解析CERS的三维结构。同时从上文的介绍中也发现CERS和疾病的联系十分密切,因此将CERS作为疾病治疗的新选择具有重大意义,但如果可以更好地了解疾病发展中CERS表达变化的分子机制,可能会更有助于疾病的治疗。
4 DES的研究进展
DES催化双键插入二氢神经酰胺,从而将二氢神经酰胺转化为神经酰胺[35](图9)。
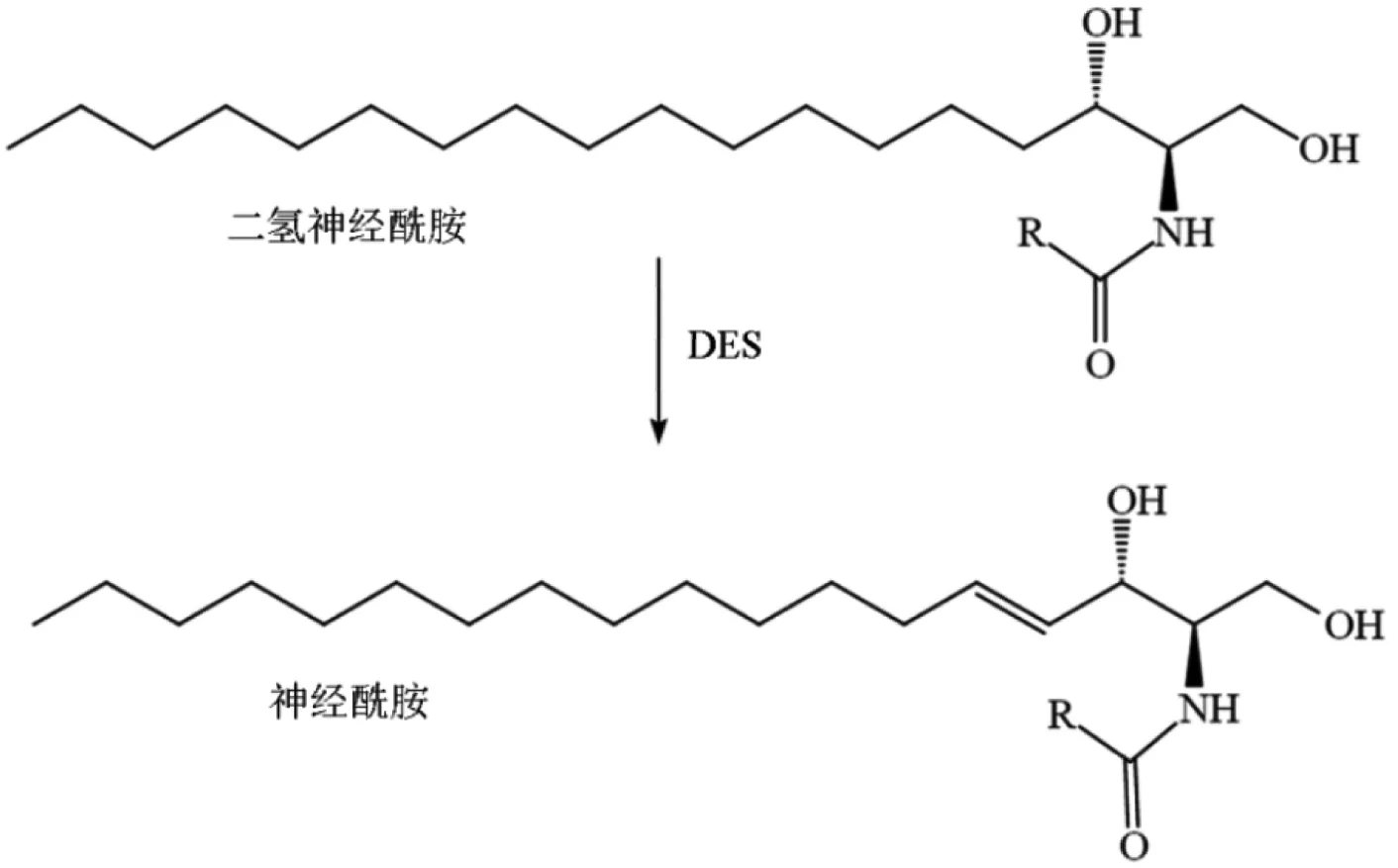
图9 二氢神经酰胺去饱和酶催化的反应[35]
DES主要包括DES1和DES2两种, DES1表现出较高的C4-去饱和酶活性和较低的C4-羟化酶活性,DES2是具有C4-去饱和酶和C4-羟化酶活性的双功能酶,目前关于DES1的研究更多。Rodriguez cuenca等[35]详细介绍了二氢神经酰胺在新陈代谢疾病中的作用;Casasampere[36]通过对DES1抑制剂的研究表明抑制DES1活性后可以防止高脂喂养后小鼠的血管功能障碍和高血压,使得该途径代表了逆转肥胖相关血管功能障碍的新靶标;Casasampere[37]研究DES1抑制剂可以通过二氢神经酰胺依赖性和非依赖性途径激活自噬,而自噬在许多疾病中均具有关键作用,因此两种途径之间的平衡最终影响细胞的命运;研究表明DES1是对抗淀粉样变性的阿尔茨海默氏病的重要治疗靶标[38]。关于DES1的最新研究进展是发表在Science上的一项研究[39],通过阻止小鼠体内DES1的活性可以减少体内神经酰胺总量,改善脂肪变性和胰岛素抵抗,从而改变代谢疾病发展轨迹,并将DES1定位成一个“可用药”靶点,对前驱糖尿病、糖尿病和心脏病这些波及数十亿人口健康的疾病具有深远的影响。
DES1的抑制剂主要包括酚类化合物和鞘脂类似物:维甲酰酚胺(4-HPR)是全反式维甲酸(一种维生素A类似物)的合成衍生物[40],是第一种抑制DES1的化学治疗药物;白藜芦醇(3,5,4′-三羟基-反式-二苯乙烯)是一种膳食多酚,可轻度抑制DES1活性[41];四氢大麻酚(THC)是大麻的主要生物活性成分[42],可以抑制大鼠肝微粒体中的DES1;化合物GT11(也称为C8-环丙烯基神经酰胺)是报道的第一个DES1抑制剂[43]。基于DES1脱饱和的机理,我们设计了一种名为XM462的5-噻二氢神经酰胺[44],XM462已被用作药理学工具,用于显示dhCer在人胃癌细胞系HGC27中作为自噬诱导剂的作用。
从近几年的研究中可以看出DES1在体内起消极作用,即抑制DES1的活性才可以对机体健康产生积极影响,因此DES1可以被定位成多种疾病的治疗靶点。如果我们可以在现有的基础上获得其三维结构,之后就可以研究对其发挥酶活重要的一些位点,可以更针对性地设计治疗疾病的药物,对疾病的靶向治疗将是重大突破。
5 总结与展望
通过对神经酰胺从头合成途径的综述,我们发现虽然研究人员已经证明了该途径在体内代谢中的重要性,可是由于途径中所涉及的酶均是膜结合的蛋白,所以除了SPT之外,其它3种酶的三维结构信息均极度缺乏,这也导致了我们对相关疾病的致病机制了解得不够透彻。如果想对它们的致病性进行深入研究或者为疾病治疗开发设计新药物,三维结构信息是必不可少的,这也为今后的研究提供了一个方向。
本文主要对神经酰胺从头合成途径疾病相关性的研究进展进行了综述,SPT突变致使功能异常导致HSAN1,KDSR功能异常主要导致PSEK,6种CERS由于其分布组织的不同,表达异常也会导致不同类型的疾病,DES表达异常也导致了多种代谢疾病。可以发现神经酰胺从头合成途径一旦发生异常,便会对个体健康产生极其严重的影响,同时目前的研究主要集中在病理蛋白层面,但是病理信号是如何逐级放大到组织层面乃至机体层面尚不清楚,这表明了对该途径进行深入研究的必要性,如可以利用蛋白质组学以及脂质组学对该途径所涉及的酶蛋白以及代谢途径进行研究。
