非晶态聚合物玻璃化转变温度的测定方法综述*
2020-08-12常伟伟王丽娜
常伟伟,王丽娜
(山东理工大学分析测试中心,山东 淄博 255000)
非晶态或部分结晶聚合物在不同温度下,表现出不同的状态和性质,除了可以呈现为液态或玻璃态外,由于其主链能通过碳碳键的内旋转运动,在特定温度下还可呈现出高弹态。非晶态或部分结晶聚合物中的非晶相从玻璃态向高弹态或橡胶态转变的温度,称为玻璃化转变温度,用Tg表示。当材料在玻璃态时,分子链和链断都不能运动,材料呈刚性。温度升到Tg以上时,分子链段之间有类似于液体的相对运动,但不足以激发整个分子链的运动,它又有固体的特点。当高聚物发生玻璃化转变时,其物理和力学性质都会发生明显的变化,如聚合物的比热、形变、粘度、折光率、质子基团运动频率等都会发生突变,对材料的储存、加工和实际应用有着重要影响[1],根据这些性质随温度的变化,可测定聚合物的玻璃化转变温度。常用的几种测量聚合物Tg方法主要有热分析技术和磁共振技术等。本文将综合分析各种实验方法的原理,优缺点及适用条件。
1 热分析技术
热分析技术是在程序温度下,测量材料的物理化学性质随温度变化技术的统称。根据测量的物理量的不同,测定聚合物Tg的热分析方法主要有差示扫描量热法(DSC)/调制差示扫描量热法(MDSC)/闪速差示扫描量热法(Flash DSC)、热机械分析法(TMA)、动态热机械分析法(DMA)。
1.1 差示扫描量热法/调制差示扫描量热法/闪速差示扫描量热法
差示扫描量热法(DSC)的原理是在程序温度下,测量物质与参比物间功率差或热流差,随温度或时间的变化。后来随着技术的不断改进,又发展出了调制差示扫描量热法(MDSC)和闪速差示扫描量热法(Flash DSC)。MDSC是在传统的线性变温速率的基础上,叠加一个周期性变化的变温速率,用以测量对该变化的变温信号有响应的热流,从而实现可逆热流与不可逆热流的有效分离,可以更准确的检测玻璃化转变过程中伴有焓松弛、冷结晶、固化、熔融、分解等现象的材料的Tg。Flash DSC 是以极高的升降温速率(已经超过107K/s),使温度变化的时间尺度低于分子链转变的特征时间,抑制样品结构的转变,从而研究样品处于某一特殊状态或结构下的性质,是研究亚稳态结构和结构重组过程的有力工具。DSC是基于检测材料在Tg前后比热的显著变化来确定材料的Tg的。比热(Cp)是高聚物内部分子运动能力的衡量标尺,材料发生玻璃化转变前后,分子链段的运动能力会发生阶跃性变化,引起比热的台阶式变化。高聚物的玻璃化转变是分子链段运动形态的变化过程,因此玻璃化转变温度不是某一定值,而是个温度区间。DSC法测定聚合物玻璃化转变,通常有两种方法来表示Tg:1)ICTA(国际热分析协会)规定将低温侧的基线外推与曲线拐点处切线的交点Teig(如图1所示)作为Tg,代表材料开始发生玻璃化转变的温度。2)ISO和国家标准中规定,低温侧和高温测的外推基线,分别与测试曲线的切线交点温度Teig和Tefg的平均温度Tmg(如图1所示)作为Tg,也就是材料热流变化速率最大时对应的温度。
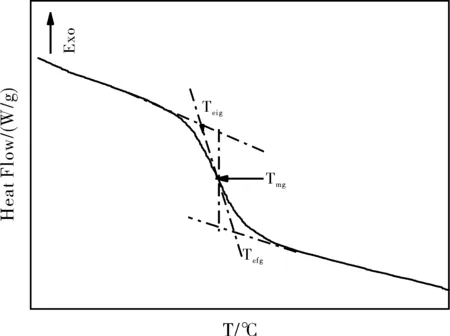
图1 测量非晶聚合物玻璃化转变温度的DSC曲线
DSC法测量聚合物Tg是发展比较早的热分析技术,实验方法比较成熟,操作简单,适合各种形态的聚合物,是聚合物Tg测试最常用的方法。在食品、化工、生物医药等领域都有广泛应用。例如,庞承焕等[2]用DSC研究了不同测试条件(包括样品用量、升温速率、裸露测试等)对铜箔基板Tg的影响。Mano等[3]用DSC研究了左旋聚乳酸的结晶度对玻璃化转变动力学和结构松弛的影响。Monnier等[4]分别用传统DSC和快速量热计对非晶体聚乳酸在其Tg以下物理老化,发现相对于传统DSC,由于快速的冷却速率使结构快速恢复,用快速量热计老化的聚乳酸增加了80%~90%的热恢复。徐丽等[5]用MDSC分析了聚苯乙烯乳液、聚苯乙烯-b-聚丙烯酸丁酯-b-聚苯乙烯、聚异戊二烯-b-聚苯乙烯与沥青混合物三种多组分体系的Tg,实验结果表明,MDSC可以有效的分离重叠的热效应,是研究多组分、复杂聚合物体系热物理性质和链段运动的有效手段。
1.2 热机械分析法
热机械分析法(TMA)在程序温度和非震动载荷作用下,测量物质的形变与温度或时间等函数关系的一种技术。聚合物在发生玻璃化转变前后膨胀系数(α)会发生明显突变,相应的材料形变也会在玻璃化转变处发生明显转折。TMA曲线在玻璃化转变前后表现为一条弧线,将该弧线前后切线的交点对应的温度,指定为Tg。相对于比热变化,尺寸变化的效应要灵敏的多,因此TMA测定聚合物的Tg要比DSC灵敏度高。Keinath等[6]用TMA的穿透模式研究了13中离子化的聚苯乙烯的Tg以及Tg以上的转变,他们发现Tg会随分子量的变化而系统的变化。Yazdi等[7]用两种TMA模式测量了聚碳酸酯(PC)/丙烯腈-丁二烯-苯乙烯共聚物(PBS)混合物的Tg,发现升温速率会影响Tg,升温速率越大,热滞后越大,此共混物只显示一个Tg。当用0.5~5 ℃/min升温时,可以检测到双玻璃化转变温度。
1.3 动态热机械分析法
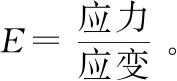
由于DMA直接测量的是聚合物的粘弹性与温度的关系,因此灵敏度比DSC高,能够检测到非常微弱的二次松弛现象。DSC无法测量的Tg,如热现象不明显或热效应有重叠的聚合物,可以用DMA测量,尤其适合高结晶、高交联的复合材料或填充材料以及嵌段/共混聚合物微弱的玻璃化转变的检测。楼倩等[9]用DMA测量了印刷电路板(PCB)材料的Tg,并研究了升温速率、测试频率和不同支架对Tg的影响。升温速率越大,测得的Tg越大。Tg值也会随着频率的增大而增大。作者还发现使用双悬臂夹具测得的Tg值明显高于使用三点弯曲测量的数值。Sun等[10]用DMA研究了商用的环氧胶黏剂和玻璃纤维增强的聚醚的热滞后对Tg的影响,实验结果显示热滞后主要有两种:一种是样品温度与仪器热电偶之间的滞后,另一种是样品长度引起的滞后。他们建立了各项同性的聚合物的模量E与温度T关系曲线,并研究了样品长度对E-T曲线和Tg的影响。
上述热分析方法中,原理不同,测得的玻璃化转变温度也不同。通常DSC得到的Tg和DMA测得的损耗模量峰值温度相差较小,TMA测得的Tg值最小。DSC是研究聚合物的热行为和检测玻璃化转变最常用、最简便的检测方法,适合各种尺寸和形态的常规样品,但灵敏度较低,对转变附近比热变化不明显或者伴随其他热效应的样品,往往得不到明显的转变信号。TMA法灵敏度较DSC高,一次实验中既可以得到玻璃化转变的信号,又可得到样品的热膨胀系数,可用于评价材料的柔韧性和短期耐热耐寒性能,但由于TMA对样品尺寸很敏感,所以对制样要求很高,要求样品表面平整,不能有气泡、开裂、填充不均等,且不适合Tg以上粘度很低的样品,不能用于粉末和半固体样品的测定。DMA在检测聚合物材料玻璃化转变和次级转变等方面,灵敏度比DSC、TMA高的多,而且得到的信息丰富,一次实验可以同时得到材料刚度、阻尼、特征温度、特征时间、特征频率等信息,在评价交联或复合材料的界面特性、共混高聚物的相容性与共混性、聚合物分子链的运动机理等方面具有实用与理论价值。DMA虽然具有更高的灵敏度,但是样品的形态、尺寸要求更严格,实验参数的设置更复杂。
2 磁共振技术
DSC、TMA、DMA等热分析技术测得的Tg值是聚合物本体的平均玻璃化转变,适用于检测物理结构和化学组成很均匀的体系。而对于食品等非均相体系以及纳米尺度的高分子材料的玻璃化转变的研究并不适用。磁共振技术可通过研究局部分子链中特征原子核或电子自旋的弛豫来表征非均相或纳米聚合物中特定链段的松弛行为、玻璃化转变行为以及链的构象等。
2.1 核磁共振波谱法
核磁共振波谱法(NMR)是将样品置于磁场中,在垂直于磁场方向,用一定频率的射频照射样品,使特定化学环境中的原子核发生共振跃迁到高能级,原子核从高能级通过非辐射弛豫回到低能级。弛豫方式有两种:(1)自旋-晶格弛豫(又称纵向弛豫)是高能级的自旋核将能量转移至周围的分子而转变为热运动。自旋-晶格弛豫时间用t1表示。(2)自旋-自旋弛豫(又称横向弛豫)指高能级原子核将能量转移给同类低能级的原子核,自旋-自旋弛豫时间通常用t2表示。NMR法测定Tg值的原理是通过测定活性原子核的弛豫特性来描述分子的运动特征。当聚合物发生玻璃化转变时,分子链段运动发生急剧变化,含有质子的基团运动频率增加,弛豫时间t1和t2变长,NMR谱线宽度变窄。这些转变对应的温度即为Tg。
NMR方法快速、灵敏,对样品没有破坏性,尤其适合食品等非均相体系Tg的测定和机理研究。Ruan等[11]用低场NMR研究了几种食品聚合物(麦芽糊精、面包、蛋糕、饼干)的Tg,发现食物在发生玻璃化转变时自旋-自旋弛豫时间t2s和自旋-晶格弛豫时间t1会发生明显变化,Tg可以很容易从双线性回归模型中得到,与用DSC或TMA检测的Tg接近,并研究了水对玻璃化转变的增塑作用。NMR还可用来研究聚合物玻璃化转变前后链段运动动力学及构型构象等变化。Zemke等[12]用多维13C NMR研究了聚苯烯、聚乙酸、聚异丙烯等几种聚合物在发生玻璃化转变时,主链的构象结构变化和动力学,揭示了γ-扭曲效应对各向同性化学位移的影响。Peng等[13]用固态NMR检测到了聚苯乙烯在玻璃化转变过程中的两步转变过程,而用DSC只能检测到第一步转变。Hu等[14]用时域1H NMR和固态13C NMR研究了聚氨酯脲弹性体的软段的玻璃化转变过程,刚性和流动性的两种链段被检测到,NMR可以很清晰的识别软段的几种重叠松弛方式。
2.2 电子自旋共振波谱法/电子顺磁共振波谱法
ESR/EPR与NMR同属于磁共振技术,ESR/EPR研究的是样品的电子磁矩与外磁场的相互作用。NMR研究的是样品核磁矩与外磁场的相互作用。由于电子磁矩远大于核磁矩,所以ESR/EPR信号灵敏度比NMR高。ESR/EPR是检测自由基等顺磁性物质最直接灵敏的方法,这是其他方法无法达到的。但对于中性的非晶态聚合物需要进行自由基标记,操作复杂。另外,ESR/EPR只能给出探针处局部结构的运动信息,对于整个链段分子的运动信息难以做出准确的判定。

表1 几种常用测试玻璃化转变温度方法比较
3 结 语
非晶或部分结晶聚合物的Tg可以通过不同的方法测定。由于依据的原理和测试条件的不同,测得的Tg也会有所变化。各种测试方法都有其各自的优缺点和适用条件。DSC操作简单,适用于各种形状和状态的聚合物,但是灵敏度较低。TMA和DMA灵敏度高,其中DMA灵敏度最高,能够检测到非常微弱的二次松弛现象,对检验原材料的质量、确定材料加工条件与使用条件等具有重要的实用价值。但操作相对复杂,对样品形态、尺寸要求严格,不适合粉末样品的测试。NMR和ESR适用于研究分子链段的局部运动。因此,在实际应用中,要根据样品的性质状态和研究目的选择适宜的测试方法。在报道或引用比较材料的玻璃化转变温度时,要说明使用的仪器、样品状态、具体的测试条件,保证测试结果的可溯源。
