3株猪博卡病毒的全基因遗传进化及重组分析
2020-08-11覃绍敏王浩刘金凤莫模双秦树英陈凤莲马玲白安斌吴健敏
覃绍敏 王浩 刘金凤 莫模双 秦树英 陈凤莲 马玲 白安斌 吴健敏

摘要:【目的】了解豬博卡病毒(Porcine bocavirus,PBoV)在广西猪群中的流行状况及遗传学特征,为有效防控PBoV感染提供科学依据。【方法】采用PCR对采自广西境内养殖场的388份猪源样品进行PBoV检测,挑选3份阳性样品(1份为PBoVG1阳性样品,2份为PBoVG3阳性样品)进行全基因扩增,测序获得的序列使用LaserGene进行处理及多重比对分析,应用MEGA 5.2中的ClustalW进行遗传进化分析,通过邻接法(NJ)构建系统发育进化树,并以SimPlot 3.5.1进行潜在基因重组事件分析。【结果】猪内脏组织(肺脏、脾脏和淋巴结的混合样品)、扁桃体、脑组织和公猪精液的PBoV阳性率分别为25.19%、8.33%、2.94%和1.54%。在所有检测样品中,PBoVG3基因群的阳性率最高,为9.02%,PBoVG2和PBoVG1基因群的阳性率均为1.55%;不同基因群的PBoV存在混合感染现象,其中,PBoVG1+PBoVG3二重感染率为1.03%,PBoVG2+PBoVG3二重感染率为0.77%。从PBoV阳性样品中扩增获得3条PBoV全基因序列(毒株),命名为GXBH2014、GXBH2015和GXLC2014,对应的GenBanK登录号为MN747332、MN747333和MN747339。其中,GXBH2014株序列全长5055 bp,GXBH2015株序列全长5070 bp,GXLC2014株序列全长4313 bp。GXBH2014株和GXBH2015株与PBoVG3基因群参考毒株SH20F各编码基因的推导氨基酸序列同源性较高,为70.9%~98.5%;GXLC2014株与PBoVG1基因群参考毒株SX各编码基因的推导氨基酸序列同源性最高,为98.0%~99.1%。基于PBoV全基因编码区(CDS)全长序列构建的系统发育进化树也显示,GXBH2014株和GXBH2015株同处于PBoVG3基因群分支上,GXLC2014株处于PBoVG1基因群分支上。基因重组分析结果显示,在GXBH2014株的全基因序列中检测到1个潜在的重组断点,位于NP1基因的第391位核苷酸;而在GXBH2015株和GXLC2014株的全基因序列中均未检测到潜在的重组断点。【结论】广西猪群中普遍存在PBoV感染,且PBoVG1、PBoVG2和PBoVG3基因群毒株同时存在。其中,GXBH2014株可能是IA159-2株(KF025386)与MN154-1株(KF025384)基因重组进化的产物。
关键词: 猪博卡病毒;全基因组;遗传进化分析;基因重组分析
Abstract:【Objective】The study was performed to survey the prevalence and genetic characterization of porcine bocavirus(PBoV) in Guangxi in order to provide scientific basis for effective prevention and control of PBoV infection.【Method】Three hundred and eighty eight porcine samples were collected from pig farms in Guangxi and were detected for PBoV by PCR. The genomic DNA of PBoVs were amplified, cloned and sequenced from three PCR positive samples(one PBoVG1 positive sample and two PBoVG3 positive samples). Multiple comparative analysis were conducted for obtained genomic sequences using LaserGene software. Genetic analysis were performed by ClustalW method using MEGA 5.2 software and neihgbor-joining(NJ) phylogenetic trees were constructed. Recombinant analysis were carried out using SimPlot 3.5.1 software. 【Result】The positive rate of PBoV infection were 25.19% in the mixture(lung, spleen and lymph node) samples, 8.33% in the tonsil samples, 2.94% in the brain samples and 1.54% in the semen samples. All three PBoV genogroups were detected in the investigated samples with the infection rate of 9.02% for PBoVG3(the highest), 1.55% for both G2 and G1, 1.03% for co-infection of G1+G3, and 0.77% for co-infection of G2+G3. Additionally, the whole genomes sequences of three PBoV strains were obtained by amplification from PBoV positive samples and were designated as GXBH2014, GXBH2015 and GXLC2014 with genomic length of 5055, 5070 and 4313 bp, respectively. The genomic sequences of PBoV strains GXBH2014, GXBH2015 and GXLC2014 were submitted to GenBank under accession numbers MN747332, MN747333 and MN747339. The deduced amino acid of GXBH2014 and GXBH2015 shared high homology with encoded genes of PBoVG3 genetic group reference strain SH20F, being 70.9%-98.5%. GXLC2014 and PBoVG1 genetic group reference strain SX shared maximum deduced amino acid identities, being 98.0%-99.1%. Phylogenetic trees based on the full-length coding sequence(CDS) of PBoV showed that PBoV strains GXBH2014 and GXBH2015 belonged to PBoVG3 genetic group and GXLC2014 belonged to PBoVG1 genetic group. Recombinant analysis showed that a potential recombinant breakpoint in NP1 gene(nucleotide number 391) of PBoV strain GXBH2014 was identified, and no recombinant breakpoint was found in genomic sequences of PBoV strains GXBH2015 and GXLC2014.【Conclusion】Widespread infection of PBoV in swine herds in Guangxi have been observed, and genetic group strains PBoVG1, PBoVG2 and PBoVG3 exist at the same time. GXBH2014 could be a recombinant derived from its parent strains IA159-2 (KF025386) and MN154-1 (KF025384).
Key words: porcine bocavirus; whole genome; genetic evolution analysis; gene recombination analysis
0 引言
【研究意义】猪博卡病毒(Porcine bocavirus,PBoV)是一种新发现病毒,于2009年由瑞典科学家在患仔猪断奶多系统衰竭综合征的病猪体内首次发现(Blomstrom et al.,2009),随后在我国、美国及加拿大等国家相继报道猪群中普遍存在PBoV感染(Zhou et al.,2014;Zhang et al.,2015)。目前尚未筛选获得适合PBoV体外培养的传代细胞系,其致病性还不明确,但有不少学者推测PBoV与猪的呼吸道疾病和腹泻相关疾病有关(Zhou et al.,2014)。因此,开展PBoV分子流行病学调查及其遗传学分析,对有效防控PboV感染具有重要意义。【前人研究进展】PBoV为无囊膜、二十面体的单股线性DNA病毒,属于细小病毒科(Parvoviridae)博卡细小病毒属(Bocaparvovirus)成员,其基因组约5 kb,编码区包括3个开放阅读框(ORF):ORF1、ORF2和ORF3(Lau et al.,2011;穆乐等,2020)。其中,ORF1位于基因组5'端,编码非结构蛋白NS1;ORF2位于基因组3'端,编码2个结构蛋白,即衣壳蛋白VP1/VP2,VP1包括整个VP2;ORF3位于ORF1和ORF2之间,编码非结构蛋白NP1(Jiang et al.,2014)。PBoV具有高度的遗传多样性,通过遗传进化分析可将其划分为G1、G2和G3等3个基因群(覃绍敏等,2014;Zheng et al.,2016)。基因重组是病毒进化的一个重要机制,通过基因重组可产生大量的遗传变异。博卡病毒(Bocavirus,BoV)也不例外,在所有的BoV中,人源博卡病毒(HBoV)的基因重组现象最早被发现。Kapoor等(2010)认为基因重组在HBoV不同编码基因中均能发生;Fu等(2011)报道HBoV2是由HBoV1与HBoV4发生基因重组而产生;崔换弟等(2014)则认为HBoV1~HBoV4间存在基因重组关系,HBoV3可能是由HBoV1与HBoV4重组进化而来,HBoV4可能是由HBoV2与HBoV3重组进化而来。有关PBoV的基因重组现象,Lau等(2011)通过研究PBoV SH14F株VP1基因发生的基因重组事件,鉴定出2个重组断点,分别位于VP1基因的第1170和1780位核苷酸附近;Yang等(2012)在PBoV WUH1株和H18株的NP1基因中检测到可能存在基因重组; Zhou等(2018b)在PBoV GD23株中发现2个基因重组断点,重组区域位于NS1基因的第925~1959位核苷酸,主要亲本毒株为IA159-1(KF025385)株,次要亲本毒株是IN109-2(KF025383)株。【本研究切入点】在现有的研究报道中,主要采用单一的PCR对临床样品进行PBoV检测(Zhou et al.,2014),极易出現漏检,而同时采用PBoVG1/PBoVG2/PBoVG3多基因群PCR开展PBoV流行监测的研究较少,针对PBoV全基因进行重组进化分析的研究也鲜见报道。【拟解决的关键问题】采集广西猪群不同组织样品开展PBoV检测,扩增PBoV全基因序列并进行遗传进化和基因重组分析,旨在了解广西PBoV的流行状况及遗传学特征,为有效防控PBoV感染提供科学依据。
1 材料与方法
1. 1 试验材料
2009—2018年,在广西境内的养殖场采集猪源样品388份,包括内脏组织(每份样品为同一头猪肺脏、脾脏和淋巴结的混合样品)135份、脑组织34份、扁桃体24份和公猪精液195份,其中135份内脏组织和12份脑组织样品采自临床发病猪群,其余样品采自外观正常猪群。病毒DNA抽提试剂盒、DNA凝胶回收试剂盒及质粒提取试剂盒购自天根生化科技(北京)有限公司;Ex Taq DNA聚合酶、LA Taq DNA聚合酶、限制性内切酶、pMD18-T载体、dNTPs及DNA Marker购自TaKaRa公司。
1. 2 引物设计与合成
参照Zheng等(2016)的研究方法,选用PBoVG1/PBoVG2/PBoVG3 PCR扩增引物用于PBoV检测;同时以Jiang等(2014)报道的引物进行PBoV全基因扩增。所有引物均委托上海美吉生物医药科技有限公司合成,具体引物序列信息详见表1。
1. 3 病毒DNA提取
取组织样品充分研磨后加入稀释液混匀,8000 r/min离心5 min,取上清液200.0 μL,病毒DNA提取参照病毒DNA抽提试剂盒说明进行操作;取公猪精液样品200.0 μL直接用于病毒DNA提取。
1. 4 PCR扩增
参照Jiang等(2014)、Zheng等(2016)的方法进行PCR扩增。 PBoVG1/PBoVG2/PBoVG3检测PCR反应体系25.0 μL:DNA模板2.0 μL,10×Ex Taq Buffer(含MgCl2)2.5 μL,dNTPs(2.5 mmol/L)1.2 μL,上、下游引物(10.0 μmol/L)各0.25 μL,Ex Taq DNA聚合酶(5 U/μL)0.3 μL,ddH2O补足至25.0 μL。扩增程序:95 ℃预变性5 min;94 ℃ 30 s,56 ℃ 30 s,72 ℃ 30 s,进行35个循环;72 ℃延伸10 min。PBoV全基因扩增PCR反应体系25.0 μL:DNA模板2.0 μL,2×LA Taq Buffer(含MgCl2)12.5 μL,dNTPs(2.5 mmol/L)4.0 μL,上、下游引物(10.0 μmol/L)各0.5 μL,LA Taq DNA聚合酶(5 U/μL)0.25 μL,ddH2O补足至25.0 μL。扩增程序:95 ℃预变性5 min;95 ℃ 30 s,55 ℃ 30 s,72 ℃ 2 min,进行40个循环;72 ℃延伸10 min。PCR扩增产物均以1.0%琼脂糖凝胶电泳进行检测。
1. 5 测序分析
选取部分PBoV检测阳性DNA样品用于全基因扩增及测序分析。PCR扩增产物经DNA凝胶回收试剂盒进行目的条带纯化,胶回收产物与pMD18-T载体连接,再转化大肠杆菌DH5α感受态细胞,通过蓝白斑筛选出阳性菌落,抽提重组质粒进行酶切鉴定和PCR鉴定,将鉴定正确的重组质粒送至上海美吉生物医药科技有限公司进行测序。测序获得的序列使用LaserGene进行处理及多重比对分析;应用MEGA 5.2中的ClustalW进行遗传进化分析,通过邻接法(Neighbor-joining,NJ)构建系统发育进化树,Bootstrap值设为1000个重复;并以SimPlot 3.5.1进行潜在基因重组事件分析(Lole et al.,1999)。PBoV参考毒株信息见表2。
2 结果与分析
2. 1 PBoV检测结果
PCR扩增产物以1.0%琼脂糖凝胶电泳进行检测,结果显示成功扩增获得约300 bp/400 bp/500 bp的目的条带(图1),与预期结果一致。不同种类样品的PBoV检测结果显示,内脏组织的阳性率最高,为25.19%(34/135);其次是扁桃体,阳性率为8.33%(2/24);脑组织和公猪精液的阳性率均较低,分别为2.94%(1/34)和1.54%(3/195),说明PBoV在猪内脏组织中的组织嗜性明显高于扁桃体、脑组织和公猪精液。在所有检测样品中,PBoVG3基因群的阳性率最高,为9.02%(35/388),PBoVG2和PBoVG1基因群的阳性率均为1.55%(6/388);检测结果还显示不同基因群的PBoV存在混合感染现象,其中,PBoVG1+PBoVG3二重感染率为1.03%(4/388),PBoVG2+PBoVG3二重感染率为0.77%(3/388)。所有检测样品的PBoV总阳性率为10.31%(40/388),具体检测结果见表3。
2. 2 PBoV全基因组扩增与测序分析结果
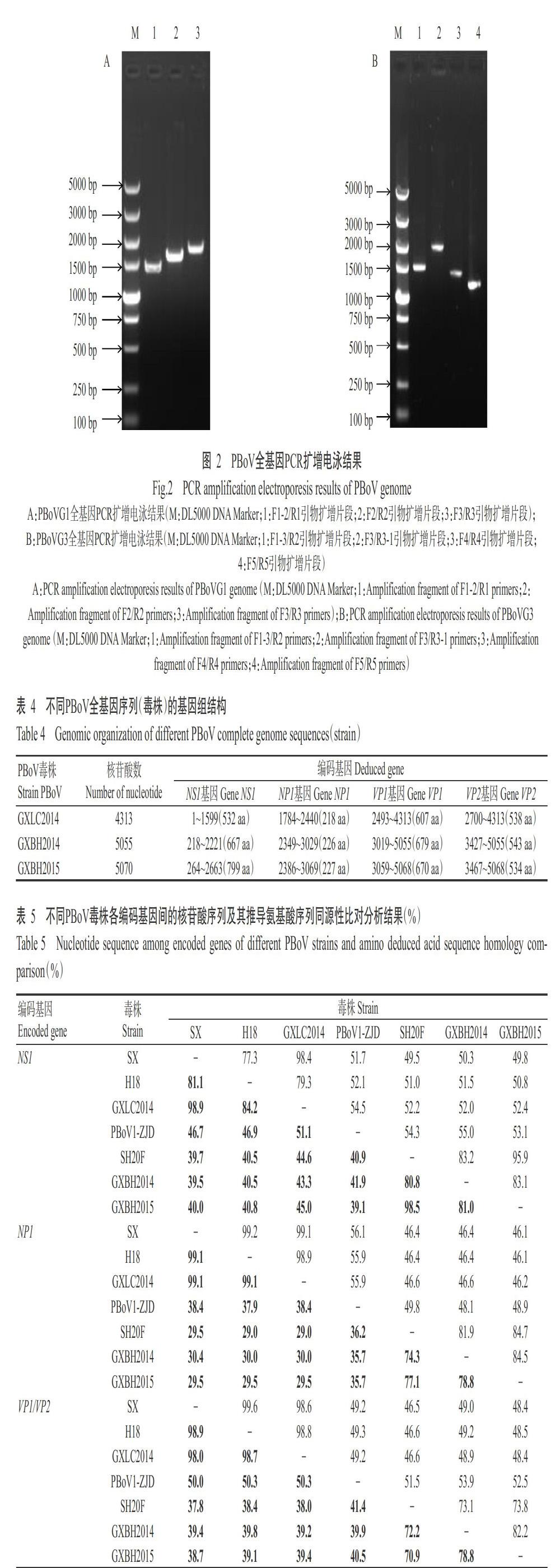
挑选3份PBoV检测呈阳性的内脏组织样品(1份为PBoVG1阳性样品,2份为PBoVG3阳性样品)进行全基因扩增,其中,1份内脏组织样品采用PBoVG1全基因扩增引物F1-2/R1、F2/R2和F3/R3进行PCR扩增,分别获得约1400、1600和1800 bp的条带(图2-A),另外2份内脏组织样品采用PBoVG3全基因扩增引物F1-3/R2、F3/R3-1、F4/R4和F5/R5進行PCR扩增,分别获得约1500、1900、1300和1100 bp的条带(图2-B),与预期结果一致。通过测序及比对分析,结果获得3条PBoV全基因序列(毒株),分别命名为GXBH2014、GXBH2015和GXLC2014,将其登录GenBank后分别获得登录号MN747332、MN747333和MN747339。其中,GXBH2014株序列全长5055 bp,包括ORF1(nt 218~2221)、ORF2(nt 3019~5055)和ORF3(nt 2349~3029);GXBH2015株序列全长5070 bp,包括ORF1(nt 264~2663)、ORF2(nt 3059~5068)和ORF3(nt 2386~3069);GXLC2014株序列全长4313 bp,包括ORF1(nt 1~1599)、ORF2(nt 2493~4313)和ORF3(nt 1784~2440)。3条PBoV全基因序列的基因组结构详见表4。
2. 3 不同PBoV毒株间的同源性分析结果
在获得的3条PBoV全基因序列(毒株)间,GXBH2014株与GXBH2015株的同源性较高,二者的NS1基因、NP1基因和VP1/VP2基因核苷酸序列同源性分别为83.1%、84.7%和82.2%,对应的推导氨基酸序列同源性分别为81.0%、78.8%和78.8%;但GXBH2014株和GXBH2015株与GXLC2014株各编码基因间的核苷酸序列及其推导氨基酸序列同源性均低于55.0%。GXBH2014株、GXBH2015株和GXLC2014株与各基因群参考毒株分别进行同源性比对分析,结果显示,GXBH2014株和GXBH2015株与PBoVG3基因群参考毒株SH20F各编码基因的推导氨基酸序列同源性较高,为70.9%~98.5%;GXLC2014株与PBoVG1基因群参考毒株SX各编码基因的推导氨基酸序列同源性最高,为98.0%~99.1%。各毒株间的同源性分析结果见表5。
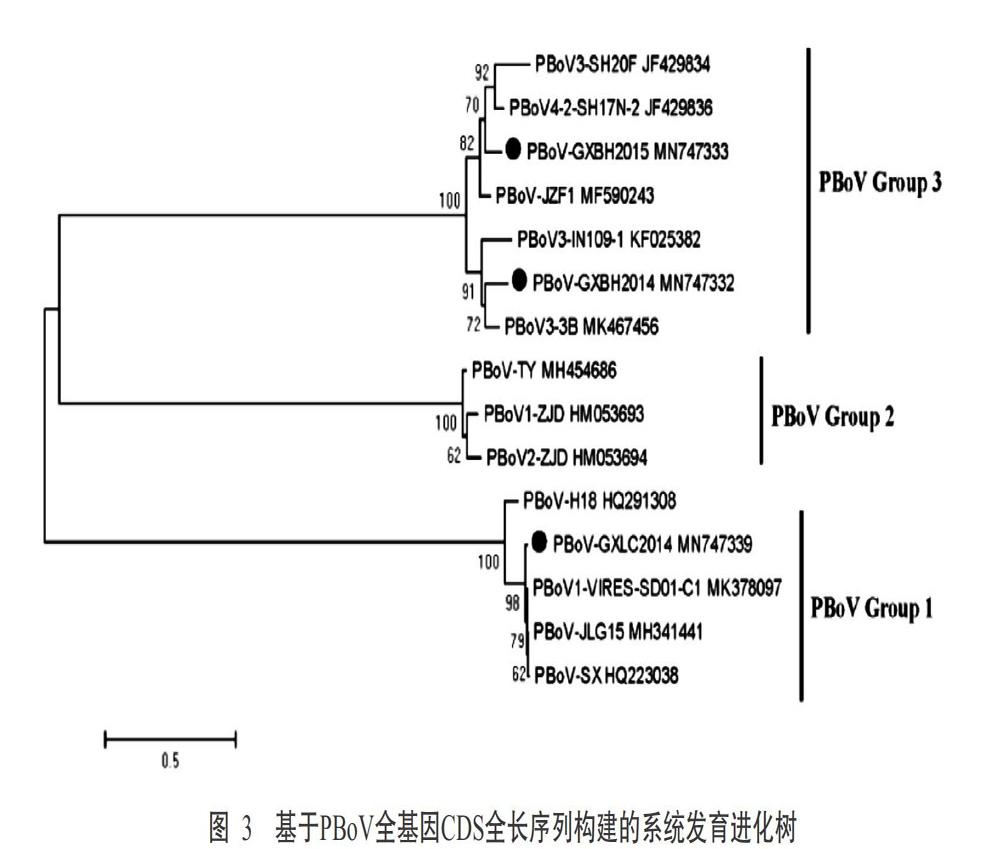
2. 4 不同PBoV毒株间的遗传进化分析结果
基于PBoV全基因编码区(Coding sequence,CDS)全长序列构建的系统发育进化树(图3)显示,所有PBoV毒株可分为三大分支,GXBH2014株和GXBH2015株同处于PBoVG3基因群分支上,其中,GXBH2014株与2013年马来西亚3B株(MK467456)的亲缘关系最近,GXBH2015株与2007年中国香港SH17N-2株(JF429836)的亲缘关系最近;GXLC2014株处于PBoVG1基因群分支上,与2017年中国VIRES-SD01-C1株(MK378097)的亲缘关系最近。
2. 5 不同PBoV毒株间的全基因序列重组分析结果
采用SimPlot 3.5.1对GXBH2014株、GXBH2015株和GXLC2014株的全基因序列进行重组分析,通过与参考毒株的全基因序列进行相似性分析并作图,结果在GXBH2014株中检测到1个潜在的重组信号(图4-A),重组断点位于GXBH2014株CDS全长序列的第2522位核苷酸,即NP1基因的第391位核苷酸,由2011年美国的2株毒株[IA159-2株(KF025386)和MN154-1株(KF025384)]重组而成。遗传进化分析结果显示,GXBH2014株NS1基因、NP1基因的第1~390位核苷酸(nt 1~390)与IA159-2株的遗传关系最近;而NP1基因的第391~681位核苷酸(nt 391~681)、VP1基因与MN154-1株的遗传关系最近(图4-B)。综上所述,GXBH2014株可能是由IA159-2株与MN154-1株发生基因重组而产生。GXBH2015株和GXLC2014株的全基因序列中均未检测到潜在的重组断点。
3 讨论
不同PBoV基因群(PBoVG1、PBoVG2和PBoVG3)间的核苷酸同源性均低于60.0%,具有高度的遗传多样性,由于缺乏共同的保守区域,无法设计可用于所有基因群毒株PCR扩增鉴定的通用引物,因此,本研究采用PBoVG1、PBoVG2和PBoVG3共3对PCR引物分别对同份样品进行检测,以避免造成漏检,或出现假阴性。在所有的检测样品中,PBoVG3基因群的阳性率为9.02%(35/388),PBoVG2和PBoVG1的阳性率均为1.55%(6/388),说明PBoVG3是广西PBoV流行的主要基因群。此外,不同基因群的PBoV存在混合感染现象,PBoVG1+PBoVG3二重感染率为1.03%(4/388),PBoVG2+PBoVG3二重感染率为0.77%(3/388),混合感染极易造成不同流行毒株间发生基因重组,可能是PBoV具有高度遗传多样性的主要原因之一。2015年,德国科学家在患脑脊髓炎仔猪的脑组织中检测到PBoV感染,且未获得其他病毒的基因组序列(Pfankuche et al.,2016)。该报道为证实PBoV具有致病性提供了有力证据,应引起研究人員的重视,并加强PBoV流行监测和致病机理研究。本研究采集猪脑组织和公猪精液样品进行PBoV检测,对应的阳性率分别为2.94%(1/34)和1.54%(3/195)。在现有的相关研究报道中,已从不同猪源样品中检测出PBoV,包括淋巴结、肝脏、肺脏、脾脏、扁桃体、血清、咽拭子、粪便及唾液等(Zhou et al.,2014;Zhou et al.,2018a;覃绍敏等,2020),由于易采集和高检出率,咽拭子和肛门拭子或粪便被公认为是开展PBoV流行监测的首选样品种类(Lau et al.,2011)。本研究从猪脑组织和公猪精液样品中检测出PBoV,表明PBoV可能具有通过猪血脑屏障和血睾屏障的能力,因此,亟待加强PBoV的流行病学及病原学研究。但由于本研究采集的样本数量有限,且脑组织和公猪精液的检出率较低,今后有必要开展更多的流行病学监测。
综合本研究的PBoVG1/PBoVG2/PBoVG3基因群检测及遗传进化分析结果可知,GXBH2014株和GXBH2015株属于PBoVG3基因群,GXLC2014株属于PBoVG1基因群。根据国际病毒分类委员会(ICTV)对PBoV的分类原则,同一个种内毒株的NS1基因推导氨基酸序列同源性>85%(Lefowitz et al.,2018),依据这一分类原则,PBoV被分为4个种,分别是有蹄博卡细小病毒2(Ungulate bocaparvovirus 2,UBoV2)~有蹄博卡细小病毒5(UBoV5),其中,UBoV2属于PBoVG2,UBoV3和UBoV4属于PBoVG1,UBoV5属于PBoVG3(Zhou et al.,2018a)。同源性分析结果显示,GXBH2014株的NS1基因推导氨基酸序列与PBoV各参考毒株的氨基酸序列同源性均低于85%,与UBoV5标准毒株SH20F的同源性最高,也仅为80.8%,因此可将GXBH2014株划分为博卡细小病毒属内的新种。本研究参照Jiang等(2014)报道的3套PCR引物(PBoV-G1、PBoV-G2和PBoV-G3)分别对PBoVG1/PBoVG2/PBoVG3阳性样品进行全基因扩增,但并未扩增获得PBoVG2的全基因序列,可能与PBoV具有高度的遗传多样性有关,仅使用1套引物不能扩增PBoVG2基因群所有毒株的全基因序列。
本研究基于PBoV全基因序列对GXBH2014株、GXBH2015株和GXLC2014株与参考毒株进行SimPlot相似性分析,当以GXBH2014株作为待分析序列、IA159-2株(KF025386)和MN154-1株(KF025384)作为参考序列时,发现参考序列的点图呈明显交叉,交叉点位于MN154-1株CDS全长序列的第2556位核苷酸,即GXBH2014株CDS全长序列的第2522位核苷酸(NP1基因的第391位核苷酸),说明可能存在重组信号;通过遗传进化分析进一步证实,在交叉点前的核苷酸序列中,GXBH2014株与IA159-2株的遗传关系最近,但在交叉点后的核苷酸序列中,GXBH2014株与MN154-1株的遗传关系最近,说明交叉点即是基因重组断点,GXBH2014株可能是由IA159-2株与MN154-1株基因重组进化而产生。已有研究证实,在HBoV不同基因型毒株(HBoV1-4)和PBoV的NS1、NP1和VP1基因中均存在重组现象(Lau et al.,2011;Yang et al.,2012;崔换弟等,2014;Zhou et al.,2018b),但有关BoV基因重组方面的研究报道较少。此外,BoV能感染犬、牛、猫、海狮、水貂、鼠及蝙蝠等多种动物(穆乐等,2020;覃绍敏等,2020),因此,加强对其他物种及各物种间基因重组的研究,对进一步了解BoV遗传学特征具有重要意义。
4 结论
广西猪群中普遍存在PBoV感染,且PBoVG1、PBoVG2和PBoVG3基因群毒株同时存在。其中,GXBH2014株可能是IA159-2株(KF025386)与MN154-1株(KF025384)基因重组进化的产物。
参考文献:
崔换弟,金玉,谢广成,程卫霞,章青,段招军. 2014. 人博卡病毒1型的近似全基因组扩增及HBoV1-4重组分析[J]. 中华实验和临床病毒学杂志,28(2):28-31. [Cui H D,Jin Y,Xie G C,Cheng W X,Zhang Q,Duan Z J. 2014. Amplification of nearly-complete sequence HBoV1 and recombination analysis among HBoV1-4[J]. Chinese Journal of Experimental and Clinical Virology,28(2):28-31.]
穆乐,孙锦涵,张健,张淋然,丁彦琴,孙玉宁. 2020. 犬博卡病毒MVC结构蛋白VP1多克隆抗体制备及特异性鉴定[J/OL]. 中国动物传染病学报. http://kns.cnki.net/kcms/detail/31.2031.s.20200102.1401.002.html. [Mu L,Sun J H,Zhang J,Zhang L R,Ding Y Q,Sun Y N. 2020. Preparation and specificity identification of polyclonal antibodies against bocavirus MVC VP1 protein[J/OL]. Chinese Journal of Animal Infectious Diseases. http://kns.cnki.net/kcms/ detail/31.2031.s.20200102.1401.002. html.]
覃绍敏,王浩,刘金凤,莫模双,秦树英,陈凤莲,马玲,白安斌,吴健敏. 2020. 广西外观正常猪群中猪博卡病毒检测及全基因遗传演化分析[J/OL]. 中国动物传染病学报. http://kns.cnki.net/kcms/detail/31.2031.S.20200318.1724. 016.html. [Qin S M,Wang H,Liu J F,Mo M S,Qin S Y,Chen F L,Ma L,Bai A B,Wu J M. 2020. Prevalence and phylogenetic analysis of porcine bocavirus in asymptomatic nursery piglets in Guangxi Province[J/OL]. Chinese Journal of Animal Infectious Diseases. http://kns.cnki.net/kcms/detail/31.2031.S.20200318.1724.016.html.]
覃紹敏,吴健敏,马琳,袁龙,陈凤莲,马玲,白安斌. 2014. 猪博卡病毒全基因组序列分析与基因分型研究[J]. 中国预防兽医学报,36(2):150-153. [Qin S M,Wu J M,Ma L,Yuan L,Chen F L,Ma L,Bai A B. 2014. Analysis of complete genomic characterization and the genotyping classification of porcine bocaviruses[J]. Chinese Journal of Prevention Veterinary Medicine,36(2):150-153.]
Blomstrom A L,Belák S,Fossum C,McKillen J,Allan G,Wallgren P,Berg M. 2009. Detection of a novel porcine boca-like virus in the background of porcine circovirus type 2 induced postweaning multisystemic wasting syndrome[J]. Virus Research,146(1-2):125-129.
Fu X L,Wang X C,Ni B,Shen H X,Wang H,Zhang X D,Chen S X,Shao S H,Zhang W. 2011. Recombination analysis based on the complete genome of bocavirus[J]. Virology Journal,8:182. doi: 10.1186/1743-422X-8-182.
Jiang Y H,Xiao C T,Yin S H,Gerber P F,Halbur P G,Opriessnig T. 2014. High prevalence and genetic diversity of porcine bocaviruses in pigs in the USA,and identification of multiple novel porcine bocaviruses[J]. The Journal of General Virology,95(2):453-465.
Kapoor A,Simmonds P,Slikas E,Li L L,Bodhidatta L,Sethabutr O,Triki H,Bahri O,Oderinde B S,Baba M M,Bukbuk D N,Besser J,Bartkus J,Delwart E. 2010. Human bocaviruses are highly diverse,dispersed,recombination prone,and prevalent in enteric infections[J]. The Journal of Infectious Diseases,201(11):1633-1643.
Lau S K P,Woo P C Y,Yip C C Y,Li K S M,Fu C T Y,Huang Y,Chan K H,Yuen K Y. 2011. Co-existence of multiple strains of two novel porcine bocaviruses in the same pig,a previously undescribed phenomenon in members of the family Parvoviridae,and evidence for inter- and intra-host genetic diversity and recombination[J]. The Journal of General Virology,92(9):2047-2059.
Lefowitz E J,Dempsey D M,Hendrickson R C,Orton R J,Siddell S G,Smith D B. 2018. Virus taxonomy:The database of the International Committee on Taxonomy of Virus (ICTV)[J]. Nucleic Acids Research,46(D1):D708-D717.
Lole K S,Bollinger R C,Paranjape R S,Gadkari D,Kulkarni S S,Novak N G,Ingersoll R,Sheppard H W,Ray S C. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India,with evidence of intersubtype recombination[J]. Journal of Virology,73(1):152-160.
Pfankuche V M,Bodewes R,Hahn K,Puff C,Beineke A,Habierski A,Osterhaus A D M E,Baumg?rtner W. 2016. Porcine bocavirus infection associated with encephalomye-litis in a pig,Germany(1)[J]. Emerging Infectious Disea-ses,22(7):1310-1312.
Yang W Z,Yu J M,Li J S,Cheng W X,Huang C P,Duan Z J. 2012. Genome characterization of a novel porcine bocavirus[J]. Archives of Virology,157(11):2125-2132.
Zhang Q Z,Zhang C H,Gao M X,He X Z,Diao Y H,Goyal S M,Mor S K,Huang J H. 2015. Evolutionary,epidemiological,demographical,and geographical dissection of porcine bocavirus in China and America[J]. Virus Research,195:13-24.
Zheng X W,Liu G P,Opriessnig T,Wang Z N,Yang Z Q,Jiang Y H. 2016. Development and validation of a multiplex conventional PCR assay for simultaneous detection and grouping of porcine bocaviruses[J]. Journal of Virological Methods,236:164-169.
Zhou F,Sun H T,Wang Y Y. 2014. Porcine bocavirus:Achievements in the past five years[J]. Viruses,6(12):4946-4960.
Zhou Y,Xu J,Wang W L,Song S W,Zhu S K,Meng Q F,Yu F,Li C P,Liu N,Luan W M. 2018a. A TaqMan-based real-time PCR assay for the detection of ungulate bocaparvovirus 2[J]. Journal of Virological Methods,261:17-21.
Zhou Y,Xu J,Zhu S K,Meng Q F,Lin Z X,Chen R,Qian A D. 2018b. Genetic analysis of three porcine bocaparvoviruses and identification of a natural recombinant breakpoint in NS1[J]. Archives of Virology,163(3):707-712.
(責任编辑 兰宗宝)
