辣根过氧化物酶催化表面修饰聚酰亚胺纤维及其润湿功能性
2020-08-11李志强王发阳王士华党洪洋
李志强 王发阳 王士华,* 党洪洋 龙 柱
(1.连云港市工业投资集团有限公司,江苏连云港,222002;2.江南大学纺织科学与工程学院造纸研究室,江苏无锡,214122)
聚酰亚胺(PI)纤维作为一种新兴的有机合成纤维,近年来广泛应用于航空航天、军事、汽车及建筑行业[1-3]。PI 纤维本体是由苯环、五元杂环、酰亚胺环、羧基等结构单元形成的有序线性高分子聚合物[4-5]。PI 纤维具有突出的热稳定性,优异的机械性能,良好的电绝缘性和低介电性能[6-7]。然而,作为有机合成纤维,PI 纤维表面惰性基团较多且比表面积较小,导致纤维的润湿性较差且较难均匀分散在水介质中,大大限制了PI 纤维进一步的推广应用[8-10]。因此,对PI 纤维进行表面处理,克服其表面缺陷,对增强PI纤维亲水性能极为重要。
目前提高PI 纤维润湿性的方法更多集中于化学法和物理法,然而,这些都不可避免地会使纤维损失部分优良性能[11]。因此,近些年来,生物酶催化法由于具备催化能力强、产物纯度高以及绿色环保等优点,在改善纤维的表面活性和润湿性方面得到了较为广泛的应用。厉世能[12]使用生物酶/H2O2体系对芳纶纤维进行表面处理,在纤维表面引入甲基丙烯酸缩水甘油酯(GMA)基团,发现纤维的表面自由能得到提高,润湿性能和拉伸性能均呈现不同程度的提升,同时保持了纤维稳定的热学性能。崔丽丽等人[13]采用生物酶催化引发甲基丙烯酸甘油酯表面接枝聚对苯二甲酰对苯二胺纤维(PPTA),并通过正交实验优化反应条件,发现纤维的表面粗糙度和蠕变性能得到改善,且保持了自身的优良性能。Xu 等人[14]使用生物酶作为催化剂将壳聚糖接枝于羊毛纤维织物上,织物的亲水性、热稳定性和染色性能等均得到了不同程度的改善。综上所述,通过生物酶催化来改善PI 纤维润湿性和分散性并保持纤维出色的本体性能成为可能。
基于此,本研究选择辣根过氧化物酶(HRP)作为高效催化剂,将“固体双氧水”——过碳酸钠(SPC)作为酶氧化剂,在纤维表面产生自由基,在反相微乳液环境中通过自由基聚合使得末端带有多羟基的磷酸单酯(PMOE)生长于PI 纤维表面,改善了纤维的表面活性和亲水性能,且对纤维本体性能损伤较小,此外,借助于湿法造纸技术得到了孔径分布均匀的PI-PMOEs 纤维纸张,期望在未来生物酶催化改性在PI纤维的亲水性改善方面有更好的应用前景。
1 实 验
1.1 材料与仪器
干纺芳香族聚酰亚胺(PI)纤维(长度3 mm,直径11.3 μm,密度1.41 g/cm3,断裂强度4.38 GPa,断裂伸长率5.0%),江苏奥神新材料股份有限公司;对位芳纶浆粕(XGJP1201,密度1.47 g/cm3,长度0.3~0.6 mm,断裂强度1.94 GPa),上海杜邦(中国)控股有限公司;磷酸缓冲盐溶液(PBS,pH 值=6.9),福建厦门海标科技有限公司;辣根过氧化物酶(HRP,活性≥250 U/mg),江苏南京都莱生物技术有限公司;甲基丙烯酸羟乙酯磷酸酯(PMOE,≥96%),五氧化二磷(P2O5,AR),对苯二酚(HQ,AR),正丁醇(NBA,AR),正辛烷(C8H18,AR),十二烷基苯磺酸钠(LAS,AR),1,4-二氧六环(Diox,AR),过碳酸钠(SPC,≥98%),丙酮(CH3COCH3,AR)和乙醇(C2H6O,AR),上海国药集团化学试剂有限公司;去离子水,实验室自制;乙醇(γd=23.00 mJ/m2,γp=19.00 mJ/m2) 和去离子水 (γd=21.80 mJ/m2,γp=51.00 mJ/m2)用于纤维接触角测试。所有化学试剂未经进一步纯化直接使用。
立式标准纤维疏解器,PL28-2 型,咸阳泰思特试验设备有限公司;纸样抄片器,ZQJ1-B-Ⅱ型,陕西科技大学机械厂;X 射线光电子能谱仪,Thermo ESCALAB 250XI 型,美国赛默飞世尔公司;扫描电子显微镜,su1510 型,日本日立株式会社;原子力显微镜,Dimension ICON 型,美国 Bruker 公司;X 射线衍射仪,D2 Phaser 型,美国Bruker 公司;热重分析仪,Q500型,美国TA 仪器公司;动态接触角测量仪,DCAT-21型,德国Dataphysics公司;多孔材料孔径分析仪,CFP-1100A型,美国PMI公司。
1.2 实验方法
1.2.1 表面生长有PMOEs结构的PI纤维的制备
在超声波作用下使用丙酮处理PI 纤维1 h,除去PI 纤维表面的有机杂质后置于60℃烘箱中干燥待用,所得纤维记为PI。
为了提高相关反应原料和助剂的相容性,构建反相(W/O)微乳液体系作为反应环境,首先向烧杯中加入9 mL 正辛烷作为油相,随后加入6 g 乳化剂LAS和助表面活性剂正丁醇3 mL,最后加入23.4 mL PBS缓冲液(0.1 mol/L)作为水相,经略微震荡和玻璃棒搅拌后得到透明澄清的反相微乳液。
将2 mg HRP 溶于上述反相微乳液中,依次向备有温度计和冷凝回流装置的三口烧瓶中加入0.3 g PI纤维束、64.8 mL Diox、溶有HRP 的反相微乳液、12.8 g PMOE (10 mL),脱气1 h。随后分批加入0.45 g SPC,加料时间大于1 h,待加料完成后在带有持续搅拌作用的油浴锅中通氮气密闭反应,在70℃下反应14 h。待反应完成后通过真空抽滤法取出纤维,在索氏提取器中使用丙酮溶液回流24 h,在80℃烘箱中干燥得到产物纤维,记为PI-PMOEs-1。此外,在同等条件下,制得PI-PMOEs-2 和PI-PMOEs-3 产物纤维,分别表示PMOE 加入量为19.2 g(15 mL)和25.6 g(20 mL)。
1.2.2 PI-PMOEs短切纤维成纸的制备
选择PI-PMOEs-3 纤维作为湿法抄造过程的原料之一,以质量比为7∶3 将PI-PMOEs-3 纤维和芳纶浆粕置于立式标准纤维疏解器中高速分散15 min,然后将纤维悬浮液转移至纸样抄片器中经过真空抽滤和压榨得到定量为60 g/m2的PI-PMOEs-3纤维纸。
1.2.3 表征方式
采用X射线光电子能谱仪(XPS)分析PI-PMOEs纤维表面修饰前后的表面元素组成及官能团变化。操作电压为15 kV,使用Al作为阳极靶,单色发射源为Al-Kα。扫描电子显微镜(SEM)用于观察PI-PMOEs纤维表面微观形貌,以含银导电胶作为基面,并将金-钯薄层喷射于样品表面,所用加速电压为20 kV。通过热重分析仪对PI-PMOEs纤维的热性能进行分析,在氮气保护下以10℃/min的速度从40℃升温至900℃。
X 射线衍射仪(XRD)用于评测纤维的晶体结构变化,使用Cu 靶(λ=0.154 nm)作为发射源,扫描范围2θ=5°~90°。使用Jade软件拟合分析纤维结晶区面积并计算相对结晶度Xc(%),具体计算见式(1)。

式中,∑Ic为结晶区的衍射积分强度;∑Ia为无定形区的散射积分强度。
使用动态接触角测量仪对纤维的接触角进行测量,测得纤维对浸润液的接触角数据后以Owens-Wendt 法计算出纤维的表面自由能γf(mJ/m2)[15],具体计算见式(2)和式(3)。

式中,θ为纤维对浸润液的动态接触角,(°);为表面自由能中的色散分量,mJ/m2;为表面自由能中的极性分量,mJ/m2;下标l代表润湿液;下标f代表纤维。
使用原子力显微镜(AFM)在轻敲模式下对纤维表面进行扫描得到纤维表面三维形貌,共振频率在250~300 kHz,同时进行数值化分析得到纤维表面均方根粗糙度(Rq)和算数平均粗糙度(Ra),具体计算分别见式(4)和式(5)。

式中,N为在图像中的像素点数;i和j分别为图像中点的位置;zij为i和j所对应位置的峰高度,nm;zcp为距中心平面的高度,nm;zav为划定区域内的平均高度,nm。
使用分散度法评估纤维悬浮液的分散稳定性,测试时使用去离子水配置250 mL 质量分数为3‰的纤维悬浮液,搅拌均匀后静置24 h,观察纤维悬浮液的

式中,h0为初始纤维悬浮液的总高度,mL;h1为静置24 h 后悬浮液中上清液层的高度,mL;f为纤维分散度,%。
在纤维纸上裁取不少于5 个3 cm×3 cm 的样点,使用多孔材料孔径分析仪分析薄片纸的孔径分布状态。
2 结果与讨论
2.1 PI纤维表面生长机理
HRP 主要存在于植物辣根中,是由无色的酶蛋白和深棕色的铁卟啉结合而成的糖蛋白复合物。由于其性质稳定且具有独特的催化性能,在过氧化氢(H2O2)作用下可以实现HRP 对芳香族化合物的改性,因此作为一种酶制剂广泛应用于化学催化领域中[17-18]。图1 为PI 纤维在HRP 催化作用下表面生长PMOEs示意图。从图1可以看出,具体的反应机理如下:①链引发:过碳酸钠(SPC)是H2O2与碳酸钠的加合物,又被称为固体双氧水,在溶剂中可释放出在H2O2存在条件下,HRP 作为催化剂催化氧化PI 纤维。首先,HRP 被H2O2氧化为高活性的HRPⅠ型酶,HRP Ⅰ型酶通过夺取苯环酰胺间位上的氢进而产生一个自由电子,形成自由基(以PI·表示)。②链增长:PI·夺取PMOE 末端碳碳双键上的氢,产生PMOE 自由基(以PMOE·表示),使PMOE在PI纤维表面发生均聚反应。③链终止:PMOE单体与PI·进行接枝聚合,使PMOE 支链接枝在PI 纤维主链上,多个支链互相交联和缠结在纤维表面生长出磷酸单酯长链(PMOEs)网状结构[20-21]。
2.2 纤维表面微观形貌及结晶性能变化
PMOEs 在纤维表面生长的微观形貌如图2 所示。由图2可知,PI纤维呈现出光滑整洁的表面,这大大降低了纤维的界面活性。而PI-PMOEs 纤维显现出粗糙度不一的表面形貌,当PMOE加入量较少时,并非所有活性位点均能发生反应,均聚得到的PMOEs 长链分子质量也相对较小,仅能在纤维表面形成断断续续的“鱼鳞”状结构。随着PMOE 加入量的增加,更多的自由基发生反应,聚合反应更加强烈,得到的PMOEs 分子质量增加,分子链得以延伸,向纤维外侧生长出“花瓣”状聚集体。在物理缠绕扭结和化学聚合交联的共同作用下,多个长链发生聚集形成尺寸相对较大的“长丝”状结构,绝大部分PMOEs 长链沉降状态,纤维的分散度计算见式(6)[16]。
在纤维表面紧密相连形成致密的网状结构,提高了聚合物在纤维表面的包覆密度,剩余部分则延伸出纤维表面。
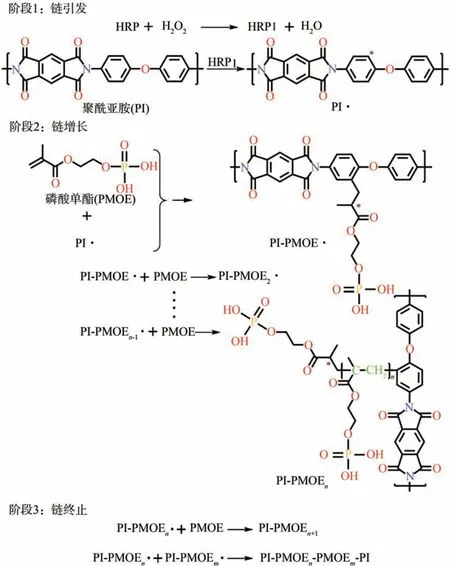
图1 HRP催化剂在PI纤维表面引发PMOE生长示意图

图2 PI和PI-PMOEs纤维的SEM图
为了进一步确认PMOEs 包覆于纤维表面,通过AFM 观察了PI-PMOEa 纤维的表面形貌三维结构如图3(a)~图3(d)所示,表面粗糙度通过Gwyddion 软件对3D 图像计算所得,结果见图3(e)。从图3 可以看出,PI 纤维表面光滑,随着PMOE 加入量增加,PIPMOEs 纤维表面更为粗糙且出现许多凸起。此外,采用均方根粗糙度(Rq)、算数平均粗糙度(Ra)和平均最大高低差(Rtm) 来评价纤维表面粗糙度。PMOE 加入量为10 mL 时,纤维表面粗糙度便得到很大提升,纤维的Ra和Rq分别由36 nm 和53 nm 增至180 nm 和223 nm,纤维表面出现的缠结长链及聚集颗粒使得Rtm提高了197 nm。当PMOE加入量为20 mL时,Ra和Rq分别增加至326 nm 和411 nm,Rtm也达到634 nm。较高的Rtm值说明纤维表面出现了很多PMOEs 长链,聚合长链向外延伸得更长,有助于提高纤维的粗糙度。此外,这些微观形貌也证明了纤维表面生长的PMOEs 结构可以通过调整PMOE 的加入量进行调控。
为了分析纤维的晶体结构变化,对PI 纤维和PIPMOEs 纤维进行了X 射线衍射分析,并计算出相对结晶度如图4 所示。从图4 可以看出,PI 纤维无定形区较多,说明纤维为半结晶聚合物,存在一定强度的衍射峰,相对结晶度为18.63%。经HRP 催化处理后,PI-PMOEs 纤维的衍射峰峰强度略微减弱,且相对结晶度均有所降低。这主要归因于PBS缓冲液的存在使得反应溶液偏碱性,而PI 纤维的耐碱性较差,纤维表面结构受到破坏,化学小分子物质渗入纤维内部,不可避免地会残留于纤维内部且较难通过索氏抽提去除,因此会轻微破坏高分子链立体规整结构,减弱了大分子链间的紧密堆砌,进而使得相对结晶度略微降低。

图3 PI和PI-PMOEs纤维的AFM三维结构图和粗糙度分析结果
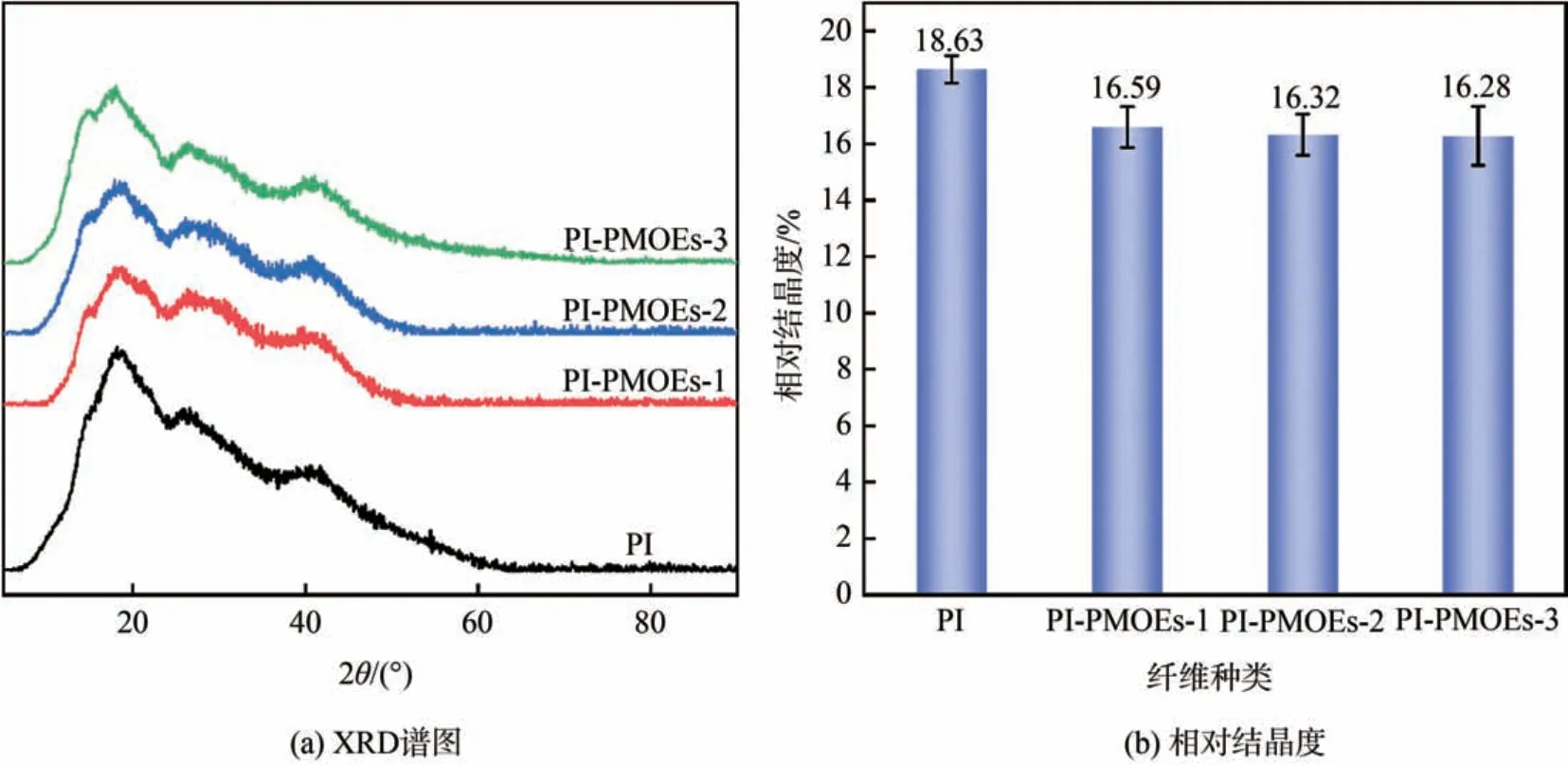
图4 PI纤维和PI-PMOEs纤维的XRD谱图和相对结晶度
2.3 纤维表面化学结构变化
使用 XPS 研究了 PI、PI-PMOEs-1、PI-PMOEs-2和PI-PMOEs-3 纤维的表面元素含量,其宽扫描测量光谱、表面不同元素和基团占比如图5 和图6 所示。由图5(a)可知,PI 纤维表面存在 C1s、N1s、O1s 峰,而PI-PMOEs纤维表面新出现了的P2p和P2s峰,选取P2p 峰作为P 元素的定量分析标准,由于纤维表面的PMOEs 中存在大量含氧基团(O—C、O=C、O=P和O—P),使得O1s 的峰强增大,而XPS 的探测深度约为10 nm,覆于纤维表面的PMOEs结构导致仅存在于纤维高分子链中N 元素的检出受限,因此N1s 峰强减弱。其他元素峰的增强致使C1s峰强度降低,这与前述的分析结果相吻合。
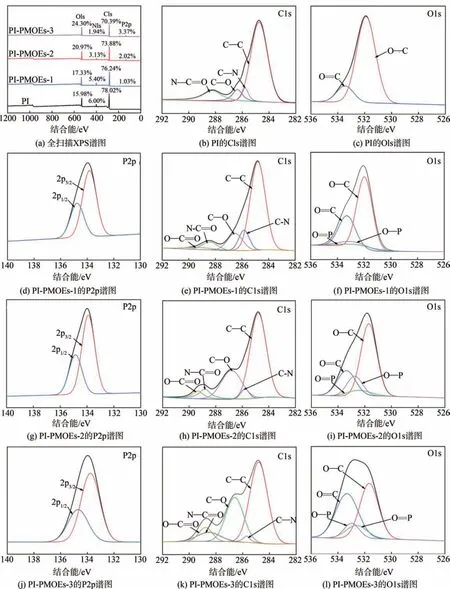
图5 PI和PI-PMOEs纤维的全扫描XPS谱图及元素谱图

图6 PI和PI-PMOEs纤维的各元素峰曲线拟合结果
此外,对各个元素峰分峰拟合的结果如图5(b)~图5(l)所示,PI 纤维的C1s 峰经拟合后可分出4 个峰,分别位于284.7 eV(C—C/C—H)、285.8 eV(C—N)、286.4 eV(C—O)和288.2 eV(N—C=O)。而PI-PMOEs 纤维的C1s 峰拟合曲线中在288.6 eV(O—C=O) 处出现新峰,如图6(a)所示,随着PMOE 加入量的增加,C—O 和O—C=O 的含量逐渐增多,分别增至29.9%和7.4%。此外,如图5(c)所示,PI 纤维的 O1s 峰仅在 531.9 eV (O—C) 和533.5 eV(O—C)处出现2 个峰,而PI-PMOEs-1 纤维的 O1s 峰分别在 532.3 eV (O=P) 和 532.8 eV(O—P)处出现新峰,且PI-PMOEs-3中O=P和O—P含量分别为10.8%和19.1%,说明PMOEs 成功地生长在了PI 纤维表面(见图6(b))。同时对3 种PIPMOEs 纤维的P2p 峰进行拟合,都在133.8 eV(P1/2)和134.7 eV(P3/2)处出现P2p的裂分峰,这也进一步证明了PMOEs的存在[22-23]。
2.4 纤维热学性能变化
纤维的TGA 和DTG 曲线如图7 所示。将分析所得的热分解参数如初始降解温度(Tdi)、最大降解速率温度(Tmax)及特定温度下的残炭率(Yc)列于表1中。从图7 可以看出,PI 纤维在605.8℃处出现了最大降解速率温度,且在490.1~800.2℃存在一段单质量损失峰,这表明PI 纤维具有优异的热性能。但是,在PI-PMOEs-3 纤维的TGA 曲线中发现分别在279.6~503.5℃和480.3~798.5℃范围内出现了两组质量损失峰,第一次热分解的质量损失约为2.19%,这一段质量损失峰是纤维表面的PMOEs 包覆层分解所产生的[24-25]。这是由于PMOE 的热分解温度较低,在PIPMOEs-1 纤维和 PI-PMOEs-2 纤维表面的 PMOE 分子质量较少,未形成较完整的PMOEs 三维网状结构,因此仅在PI-PMOEs-3 纤维的TGA 曲线中出现较明显的新质量损失峰。此外,由前所述,经HRP 催化处理后纤维内部的分子链规整性受到破坏,纤维的耐热性受到一定影响,因此所有PI-PMOEs 纤维的Yc值均略微降低。

图7 氮气气氛下PI和PI-PMOEs纤维的热降解曲线
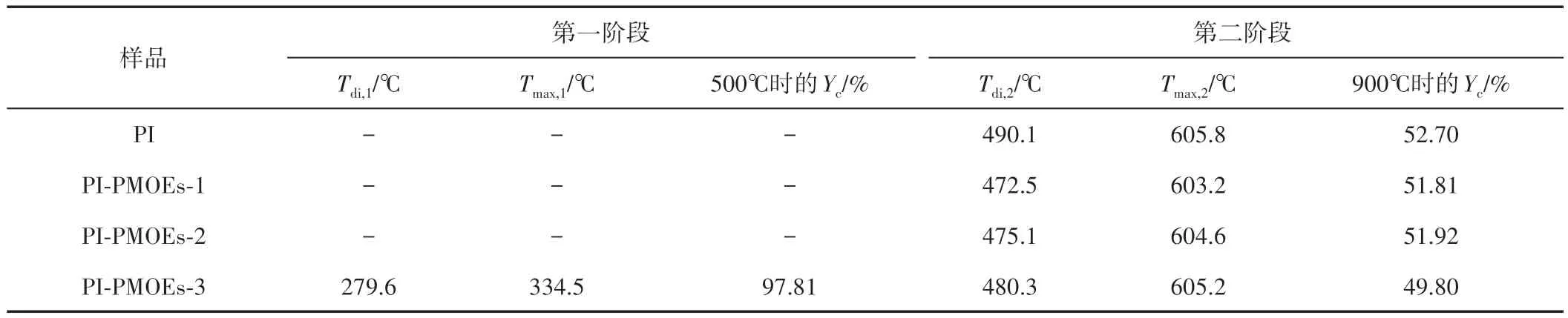
表1 氮气气氛下PI和PI-PMOEs纤维的热降解数据
2.5 纤维亲水性能变化
本研究的目的是为了提高PI 纤维的亲水性,因此表面自由能的提升有利于提高纤维表面的界面性能。测定出纤维关于去离子水和乙醇两种润湿液的接触角,同时计算出纤维表面自由能的色散分量()、极性分量() 及表面自由能(γ)f,结果如图8(a)~图8(b)所示。从图8(a)中可以看出,PI 纤维对去离子水和乙醇的接触角都相对较大,说明PI 纤维对强极性溶剂和弱极性溶剂润湿性都较差,而PIPMOEs 纤维对两种润湿液的接触角均明显减小。图8(b)中 PI-PMOEs-3 纤维的表面自由能为 42.70 mJ/m2,相对于PI 纤维的增幅达到34.36%,纤维的表面活性提高,这主要归因于纤维表面粗糙度的增加和比表面积的增大,能够促进其克服液体的表面张力,使液体更易在纤维表面铺展开来[26]。同时,可以发现PIPMOEs 纤维表面自由能的增大主要源于极性分量()的贡献,这是由于纤维表面极性、诱导效应和氢键的影响力的增强,同时极性官能团数量和种类增多,因此PI-PMOEs 纤维对去离子水的浸润性能提升程度大于其对乙醇的浸润性能提升程度,相比PI 纤维,PI-PMOEs-3 纤维对去离子水润湿液的接触角降低了13.6°,而对乙醇润湿液的接触角仅降低了9.9°。
为了更为直观地评估不同种类纤维的亲水性能,进一步测定了纤维悬浮液的分散度及纤维成纸的孔径分布,结果如图8(c)~图8(d)所示。PI 纤维的分散度为35.0%,PI-PMOEs 纤维的分散度均增大,其中PI-PMOEs-3 纤维分散度达到75.0%,相比PI 纤维增加40 个百分点。同时,经真空湿法抄纸技术得到的PI 纤维纸张中孔隙尺寸各异,而PI-PMOEs-3 纤维成纸的孔隙尺寸较小,且孔径分布较为均匀,21~50 μm 的孔径占比为57.58%,特别是21~30 μm 孔径占比达到27.53%。这是因为PI 纤维表面自由能较小,由于受到范德华力作用,纤维间会形成“棉球状”或“絮状”的纤维团,且结构紧实,一旦形成这样的结构,会产生更强烈的憎水效应,并阻碍其他纤维的分散,纤维悬浮液的分散稳定性变差,纸张中包含较多纤维束,纤维层数不均。而PI-PMOEs-3 纤维表面存在更多的极性基团,表面粗糙度增加,对水有更强的亲和力,纤维间的排斥力增强,在湿法抄造过程中,纤维束更易分散为单根纤维进而形成孔径尺寸均一的三维网状结构。总体而言,纤维的润湿性随PMOE 加入量的增加而增加,表明纤维的亲水性得到提高,有利于增强纤维的界面性能,经湿法抄造技术得到的纤维纸张匀度也更高。

图8 PI和PI-PMOEs纤维亲水性能及其成纸孔径分布
3 结 论
本研究选择辣根过氧化物酶(HRP)作为高效催化剂,将“固体双氧水”——过碳酸钠(SPC)作为酶氧化剂,在纤维表面产生自由基,在反相微乳液环境中通过自由基聚合使得末端带有多羟基的磷酸单酯(PMOE)生长于聚酰亚胺(PI)纤维表面。
3.1 通过扫描电子显微镜(SEM)和原子力显微镜(AFM)观察到PMOEs结构在PI纤维表面与之紧密相连,PI-PMOEs-3 纤维的均方根粗糙度和算术平方根粗糙度分别增至180 nm 和223 nm,并进一步通过X射线光电子能谱仪(XPS)证明了纤维表面的PMOEs层的存在且其结构可通过PMOE接枝单体加入量进行调控。说明了纤维的表面粗糙度提高,活性位点增多,有利于提高其与极性物质的亲和力及其在水介质中的分散性。
3.2 纤维的结晶性能和热稳定性能几乎未发生变化,说明生物酶催化表面修饰PI 纤维的方法对纤维的本体性能的损伤较小。
3.3 相比PI 纤维,当PMOEs 加入量为25.6 g 时,PI-PMOEs-3 纤维对去离子水和乙醇两种液体的接触角分别降低13.6°和9.9°,PI-PMOEs-3 纤维的表面自由能升至43.70 mJ/m2,且极性分量占比较大,PIPMOEs-3 纤维在水介质中的分散度也相对提高40 个百分点,说明了纤维润湿性和分散性得到大幅度改善,纤维亲水性能的提升促成了孔径分布均一的纤维成纸的抄造成形。
