近三年苏州工业园区医疗器械生产企业现场检查简析
2020-08-10钱晓明倪美华陆锃
钱晓明,倪美华,陆锃
1 苏州工业园区食品药品安全稽查大队,苏州市,215000
2 苏州工业园区市场监督管理局,苏州市,215000
0 引言
苏州工业园区是中国和新加坡两国政府间的重要合作项目,自1994年开发建设以来,已成为国内发展水平最高、竞争力最强的国家级开发区之一。生物医药是苏州工业园区聚力创新的战略性新兴产业,近年来发展迅速。截止2018年底,苏州工业园区共有医疗器械生产企业128家,2018年医疗器械产值近130亿元。实施《医疗器械生产质量管理规范》[1],确保医疗器械生产全过程的监管,对提高医疗器械产品质量至关重要,同时也是保障公众用械安全、改进监管方式、提高监管效率的重要举措[2]。我们主要结合原国家食品药品监督管理总局于2015年9月发布的《医疗器械生产质量管理规范现场检查指导原则》等4个指导原则[3-6],对自2017年1月至2019年6月的144家次苏州工业园区医疗器械生产企业质量管理规范现场检查情况进行初步统计和分析,为医疗器械生产企业质量管理体系持续改进提供参考。
1 苏州工业园区医疗器械生产质量管理规范检查情况分析
1.1 现场检查基本情况
自2017年1月至2019年6月,苏州工业园区共进行了144家次医疗器械生产质量管理规范现场检查,其中针对二类产品76家次,三类产品64家次,二类、三类同时进行有4家次。从产品类别来看,依据为《医疗器械生产质量管理规范现场检查指导原则》的有源医疗器械和软件医疗器械检查分别有62家次和7家次,分别占43%和5%;依据为《医疗器械生产质量管理规范无菌医疗器械现场检查指导原则》的无菌医疗器械检查有29家次,占20%;依据为《医疗器械生产质量管理规范体外诊断试剂现场检查指导原则》的体外诊断试剂检查有29家次,占20%;依据为《医疗器械生产质量管理规范植入性医疗器械现场检查指导原则》的植入医疗器械检查有17家次,占12%。现场检查医疗器械类型分布见表1。
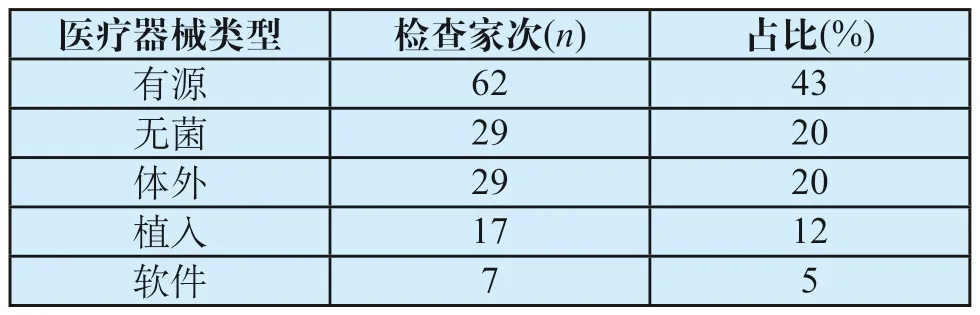
表1 现场检查医疗器械类型分布Tab.1 Medical device types distribution of on-site audits
现场检查类型分布见表2,其中注册环节现场核查85家次,占59%;生产许可现场检查33家次,占23%;注册环节与生产许可现场检查同时进行的有7家次,占5%;监督检查19家次,占13%。

表2 现场检查类型分布Tab.2 Audit types distribution
2017年1月至2019年6月的144家次现场检查中,共发现1 476条不符合项,其中,体外诊断试剂现场检查不符合项平均约为13.3条,有源和软件医疗器械不符合项平均约为9.7条(有源医疗器械和软件医疗器械的检查依据都是《医疗器械生产质量管理规范现场检查指导原则》,故两者放在一起分析),植入医疗器械检查不符合项平均约为9.5条,无菌医疗器械检查不符合项平均约为8.9条。平均不符合项数量情况见图1。可见无菌和植入医疗器械现场检查时的不符合项平均条款数量与体外诊断试剂、有源和软件医疗器械相比较少,原因是这两类医疗器械风险相对较高,企业内部对质量体系较重视,同时也是监管机构重点监管的对象,因此平均质量管理体系水平相对较好。
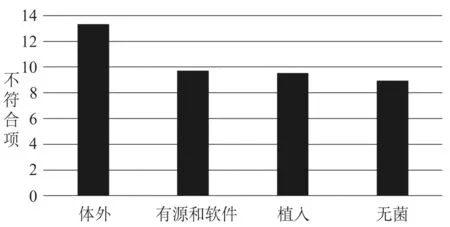
图1 平均不符合项数量情况Fig.1 Average number of findings in audits
1.2 现场检查不符合项条款分析
在所有1 476条不符合项中,出现最多的属于生产管理章节,共有不符合项219条;第二是厂房与设施章节,共有不符合项186条;第三是质量控制章节,共有不符合项185条。现场检查不符合项分布见图2。
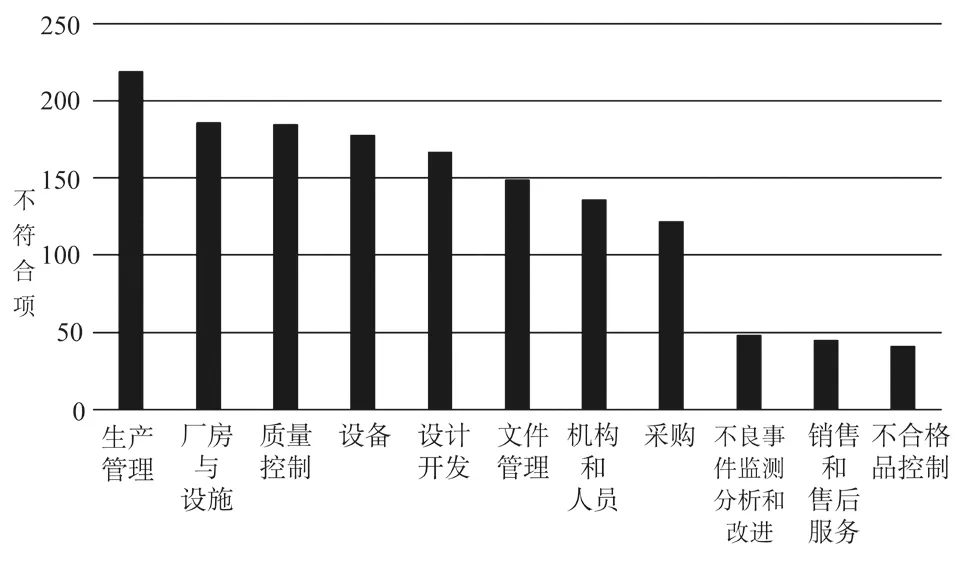
图2 现场检查不符合项分布Fig.2 Findings distribution in on-site audits
1.2.1 有源和软件医疗器械现场检查的不符合项情况
检查依据为《医疗器械生产质量管理规范现场检查指导原则》的62家次有源医疗器械和7家次软件医疗器械现场检查中,共发现了669条不符合项,最多的属于设计开发章节,共有96条不符合项;第二是文件管理章节,共有91条不符合项;第三是生产管理章节,共有78条不符合项。有源和软件医疗器械现场检查不符合项分布见图3。
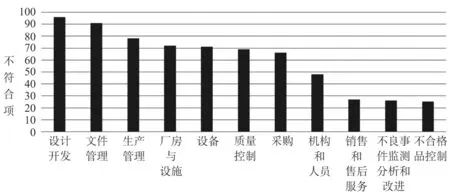
图3 有源和软件医疗器械现场检查不符合项分布Fig.3 Findings distribution in active and software medical device audits
从《医疗器械生产质量管理规范现场检查指导原则》中123条条款来看,669条不符合项最多的归于条款“7.6.2”,不符合项出现次数有38次,常见不符合如:“关键物料未记录物料料号或物料批号”“批生产记录内容不全,部分重要生产步骤或生产日期未记录”等;第二是归于条款“2.6.2”,不符合项出现次数有36次,常见不符合如:“原材料库中存放的物料没有明显的物理隔离,仓储区无退货区,召回区”“仓储区中某些物料没有放在规定的区域内,且没有明显的状态标识”“仓储区原材料、包装材料、中间品、待发货产品混放”等;第三是归于条款“8.4.2”,不符合项出现次数有30次,常见不符合如:“检验报告中未记录原始数据”“测量要求与测量结果不统一”“检验报告中缺少要求的项目”等。
1.2.2 无菌医疗器械现场检查的不符合项情况
检查依据为《医疗器械生产质量管理规范无菌医疗器械现场检查指导原则》的29家次现场检查中,共发现了259条不符合项,最多的属于质量控制章节,共有45条不符合项;第二是厂房与设施章节,共有42条不符合项;第三是生产管理章节,共有37条不符合项。无菌医疗器械现场检查不符合项分布见图4。
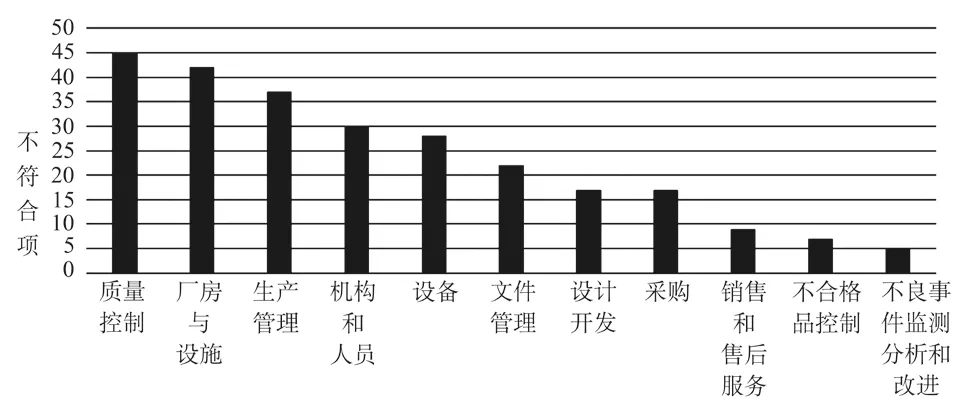
图4 无菌医疗器械现场检查不符合项分布Fig.4 Findings distribution in the sterile medical device audits
从《医疗器械生产质量管理规范无菌医疗器械现场检查指导原则》中188条条款来看,259条不符合项最多的归于条款“7.6.2”,不符合项出现次数有11次,常见不符合如:“生产记录中记录的参数与实际不一致”“生产记录中缺少主要设备的名称与工艺参数,记录不完整”“涂层工序中,要求涂层溶液在24小时内使用,但生产记录中未能体现对时间的控制”等;第二是归于条款“2.6.2”,不符合项出现次数有10次,常见不符合如:“原材料仓储区未按照待检、合格、不合格、退货或者召回进行合理划分”“无菌待检放行区,未对已抽检产品和未抽检产品进行标识区分”“成品库未设置退货区或召回区,部分产品没有设置货位卡”等;第三是归于条款“8.4.2”,不符合项出现次数有8次,常见不符合如:“部分检验设备的检验记录信息不全”“纯化水检验原始记录无原始数据,如不挥发物的测定”“进货检验记录中未填写生产厂家的原始批号”等;第四是归于条款“8.8.1”,不符合项出现次数有8次,常见不符合如:“检验用化学试液的配制记录,只有质量和体积,没有称重的原始记录”“《纯化水作业管理规定》中对纯化水的温度处理未作规定”“《工艺用水管理规定》中,未对清洗管道所用的碱液做出规定”等。
1.2.3 植入性医疗器械现场检查的不符合项情况
检查依据为《医疗器械生产质量管理规范植入性医疗器械现场检查指导原则》的17家次现场检查中,共发现了162条不符合项,最多的属于质量控制章节,共有34条不符合项;第二是生产管理章节,共有32条不符合项;第三是厂房与设施章节,共有19条不符合项。植入性医疗器械现场检查不符合项分布见图5。
从《医疗器械生产质量管理规范植入性医疗器械现场检查指导原则》中216条条款来看,162条不符合项最多的归于条款“7.6.2”,不符合项出现次数有8次,常见不符合如:“批生产记录中无涂层干燥结束时间”“生产记录中缺少对主要设备的记录,部分工序未记录具体参数,如清洗烘干时间等”“批生产记录中金属清洗工序中清洗过程参数缺少时间参数”等;第二是归于条款“8.4.2”,不符合项出现次数有5次,常见不符合如:“无菌检验抽样量与检验规范不一致”“过程检验记录中未标明量具编号,无法追溯”等;第三是归于条款“8.8.1”,不符合项出现次数有4次,常见不符合如:“缺少工艺用水量分析报告”“纯化水二级反渗透在线监测值与出水口监测值差异过大,企业未查找原因”“《工艺用水管理制度》规定品质部对工艺用水取样,《生产车间纯化水系统取样和监测程序》规定工艺部门对工艺用水取样,文件规定互相矛盾”等。
1.2.4 体外诊断试剂现场检查的不符合项情况
检查依据为《医疗器械生产质量管理规范体外诊断试剂现场检查指导原则》的29家次现场检查中,共发现了386条不符合项,最多的属于生产管理章节,共有72条不符合项;第二是设备章节,共有64条不符合项;第三是厂房与设施章节,共有53条不符合项。体外诊断试剂现场检查不符合项分布见图6。
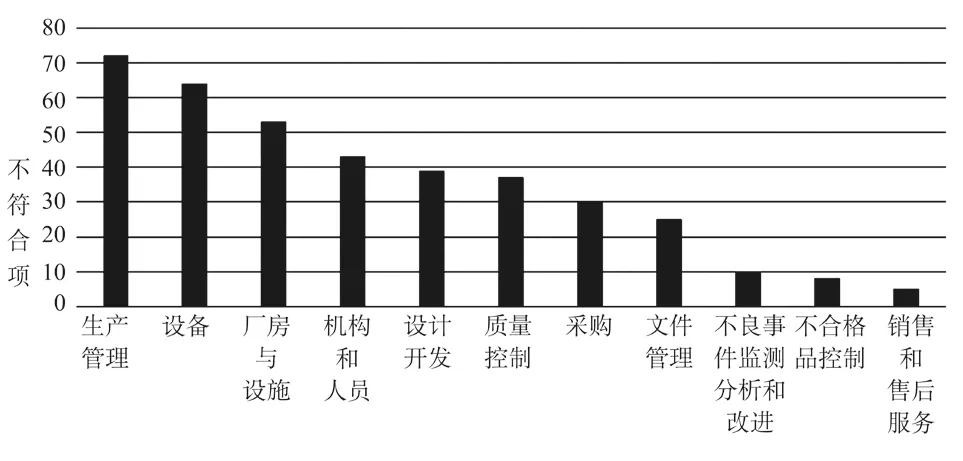
图6 体外诊断试剂现场检查不符合项分布Fig.6 Findings distribution in IVD audits
从《医疗器械生产质量管理规范体外诊断试剂现场检查指导原则》中218条条款来看,386条不符合项最多的归于条款“7.6.2”,出现次数有13次,常见不符合如:“部分工序未记录工艺参数,如自动加样机选择的程序编号等”“生产记录中缺少对主要设备的记录”“批生产记录中未见细胞裂解液原材料的领料记录”等;其次是归于条款“6.5.2”,不符合项出现次数有12次,常见不符合如:“磁珠原材料技术要求文件中的效价标准要求与磁珠的检验作业指导书标准不一致”“原材料采购记录不全,如缺少谷丙转氨酶的检验报告”“采购记录中缺少部分供应商的资质材料”等。第三是归于条款“1.12.1”,不符合项出现次数都是11次,常见不符合如:“《产品清洁管理制度》中未对洁净服清洗一次的最大数量进行规定,且清洗记录中未记录每次清洗洁净服的数量”“洁净区内待用洁净服未标注清洗日期和有效期,个别未编号”“万级洁净区工作服缺少灭菌设备,未达到灭菌要求”等;第三同时归于条款“2.6.2”,不符合项出现次数都是11次,常见不符合如:“仓储区相关标识不清晰,不合格区与包材区混放;仓库现场发现与企业无关的物品存放,且无任何标识”“货位卡上记录项目不全,不便于追溯”等。
2 讨论与分析
综合来看,苏州工业园区的医疗器械生产企业的质量管理水平参差不齐,部分质量管理水平较差的企业与高水平企业相比,人员对质量管理体系的理解不足,质量意识不强,质量体系全员参与不够,质量管理体系对产品实现贴合不紧,在以下几个方面问题比较突出:
(1)厂房与设施方面
洁净室问题,如从天花板进入洁净区的管道密封不严;洁净室需要显示压差的位置缺少压差计,或者压差计损坏未及时修理或更换;万级生产区与外界相连的传递窗无法实现互锁;水池排水管与下水口连接处未密封等。生产场地问题,如部分电子元器件的贮存不符合静电防护要求等。仓库问题,如原材料货位卡记录不全,主机板的货位卡缺少供应商信息;物料仓库无温度检测装置等。另外,很多体外诊断试剂的生产企业如果生产对应的仪器,仪器的生产会存在一系列的问题,如仪器生产区域面积偏紧,不同产品生产区域不分区;生产工序工位不明确,生产工具、原材料存放较乱,不同物料堆放在一起等等。
(2)设备方面
设备无法满足要求,如洁净区纯化水设施末端洗手池水阀为手动,易造成污染。设备放置问题,如冷库备用汽油发电机放置在过道,未设置排烟设施。设备维护保养问题,如设备管理控制程序中要求的设备保养周期和保养项目与实际操作不一致;洁净区内冷库地面欠清洁,称量间天平托盘下有粉尘等。设备管理问题,如工装未列入设备管理,尤其是工厂自制的工装,如程序烧录工装,特殊检验工装等常不作统一管理和控制。另外,很多企业对设备状态标识管理做的不到位,存在设备误用的风险。
(3)文件控制方面
文件控制方面的不符合主要有文件规定与实际操作不一致,如操作规程的实际批准人员与文件控制程序规定的不一致;不同文件规定不一致,如质量主管任命书规定的岗位职责与《质量部质量职责》规定的内容不一致,不同文件中的灭菌组批规则不一致;过期版本的操作指导书、记录表等在现场也屡见不鲜;另外涂改文件之后未签姓名和日期也很常见。
(4)设计开发方面
在设计开发方面的不符合涉及到风险管理、设计转换、设计变更等,其中设计变更问题比较突出。通过与生产企业交流来看,一是对设计变更控制节点概念不清,把握不准,导致有些企业变更记录过于简单,有些企业过于复杂。一般而言,该节点是样品验证完成,图纸发行生效,批准进入试生产的时间点。在此节点之前的变更,按照设计评审进行控制。节点之后的变更,可能涉及到供应商、模具、库存、工艺、工装等,应参照生产变更控制。二是对变更重视不足,各部门沟通不畅。变更后不记录,应该变更却不作变更时有发生。如有源设备在进行EMC测试时未通过,现场加装磁环再次测试后通过,有的企业在批量生产时直接变更,未作变更控制和记录,有的企业未及时将测试时的变更信息传递给产品开发,批量生产时就没装磁环。
(5)生产管理方面
批生产记录问题:如生产记录中重要设备的型号和参数未作记录;生产记录中缺少重要原材料的批号或者序列号。生产作业指导书与实际操作不符:如作业指导书要求在清洗完的焊接点敷上焊锡,现场检查中未发现有清洗过程和清洗设备;工艺流程及作业指导书中工艺名称与批生产记录中的不一致等。生产人员疏忽问题:如缓冲间中原材料存放区辅料库存数量与实际不符;消毒液过期;装配防静电区未接地等。
(6)质量控制方面
质量控制方面的不符合主要是检验设备未及时校验;检验时未记录原始数据,只记录合格或不合格;检验设备放置问题如电子天平,与水浴锅、磁力搅拌器摆放在同一台面,易对天平产生干扰等。
以上问题的原因,一是人员经验不足。近几年医疗器械行业在中国发展迅猛,有经验的医疗器械研发、生产、质量人员短缺。医疗器械质量体系很多借鉴了其他有经验公司的体系,在理解、转化、落实上有偏差和不足,导致不同文件之间,文件与实际操作之间存在矛盾和差距。二是全员参与不足。质量管理是公司全体人员的事情,不单单是质量部或者是管理层的事。全员参与不足造成了很多规定与实际操作的不一致,文件规定之间的矛盾,都没有在平时文件的使用过程中被发现。三是部分内审流于形式。内部审核领导不重视,计划不周全,实施不严格,整改不到位。审核员找不出问题,找到问题不深挖,问题整改浮于表面,只整改发现的问题,不解决根源问题,不考虑相关问题。
3 结语
我们仅对2017年1月至2019年6月近三年苏州工业园区内进行的产品注册、生产许可、监督检查现场检查报告内容中不符合项进行了初步统计分析,同时对出现频率较高的不符合项作了进一步分析。解决以上问题,关键在于企业需要培养和建立员工对质量管理体系持续提升和改进的意识,提升员工发现问题解决问题的能力,鼓励员工结合公司实际,对公司流程、模板、工装、设施设备等提供改进建议,使流程更顺畅,模板使用更方便,工装使用更顺手,质量体系与产品实现贴合更紧密。从而不断完善质量管理体系,保证持续稳定的生产出安全有效的医疗器械产品。
