对激光治疗设备注册后升级设计开发更改的风险控制的研究
2020-08-10官辉煜许芳傅海涛王婷张聪黄亮袁炜朱文迪湖北省药品监督管理局技术审评核查中心武汉市43007湖北省医疗器械质量监督检验研究院武汉市430075
官辉煜,许芳,傅海涛,王婷,张聪,黄亮,袁炜,朱文迪 湖北省药品监督管理局技术审评核查中心,武汉市,43007 湖北省医疗器械质量监督检验研究院,武汉市,430075
0 引言
近年来,我们现场检查时发现,企业不按照医疗器械生产质量管理规范[1]的要求更改注册产品设计的问题较为突出,部分产品与注册检验送检样机差异较大,外观、关键元器件、软件上都做过大的更改,但在设计开发文档找不到相应更改的记录。企业出于各种原因及顾虑不愿意承认注册产品发生过设计更改,对更改后产品的风险也未进行有效控制,导致产品的安全、有效性无从保证。回顾历史,国内外重大医疗器械安全事故多出于设计开发更改过程中未防控风险,因此设计开发更改对医疗器械生产过程有着举足轻重的影响。我们通过调研和现场检查等方式,系统地收集生产企业常见设计开发更改实施情况,对激光治疗仪常见设计开发更改成因及更改风险进行分析,研究关键元器件的更改及其应对控制措施,形成对激光治疗设备设计更改的识别方法,并对设计开发更改如何处理给出了建议。有助于帮助检查员从技术的角度上找准产品设计更改风险点和评估安全水平,并指导企业正确认识产品设计开发更改的重要性,及时预防可能发生的风险,进一步保证产品质量。
1 激光治疗设备的治疗原理及分类
激光治疗主要是通过激光的生物刺激、调节作用和利用焦点处的高能、高温、高压的电磁场作用和烧灼作用,对病变组织进行切割、黏合、气化。激光器按工作介质分为四类:一是气体激光器,是指采用气体激发而产生激光的激光器,常见的有He-Ne激光器、CO2激光器、准分子激光器、铜蒸气激光器等;二是固体激光器,是指采用固体发光物质为发光器件的激光器,常见的有ND:YAG激光器、红宝石激光器、翠绿宝石激光器、钬激光器、铒激光器等;三是半导体激光器,是用半导体材料作为工作物质的激光器。核心发光部分是由P型和N型半导体构成的PN结管芯,当注入PN结的少数载流子与多数载流子复合时,会发出可见光,紫外光或近红外光,激励方式有电注入、电子束激励和光泵浦三种形式;四是液体激光器,包括染料激光,以染料为介质而产生激光辐射,最重要的特征是其波长连续可调谐。
2 激光治疗设备常见设计开发更改成因
2.1 法律法规、标准或其他规定要求的发布或修订
主要包括有以下几个方面:国家食品药品监督管理总局令第6号《医疗器械说明书和标签管理规定》于2014年10月1日起施行;国家食品药品监督管理总局发布了医疗器械生产企业质量控制与成品放行指南的通告(2016年第173号);国家食品药品监督管理局办公室印发YY 0505-2012医疗器械行业标准实施工作方案的通知食药监办械(2012年第149号);激光产品有关标准的变化,如国标GB 9706.20[2]、GB 7247.1[3];行标YY 0307[4]、YY 0599[5]、YY/T 0758[6]、YY 0789[7]、YY 0844[8]、YY 0845[9]、YY 0846[10]、YY 0983[11]、YY 1289[12]、YY 1300[13]、YY 1301[14]、YY 1475[15]等。
2.2 在生产过程中发现制造、安装、服务困难过大
一是供应商发生变化,主要是停产、供货不及时、质量下降、价格问题、服务不好;二是生产效率不足,产品制造工艺过于复杂,费时费力,导致生产效率低;三是安装调试复杂,导致耗时过长。
2.3 顾客或供方要求的更改
主要更改需求包括更改产品外形、提高产品某些指标、加入特殊要求的功能等。
2.4 纠正和预防措施所要求的更改
包括事后识别出的,在设计阶段产生的计算、材料选择等错误。首先是设计开发阶段考虑不周全,关键元器件设计选型未考虑到量产的需要,批量生产产品的离散性导致部分产品性能指标不合格率高;其次是管理评审和内部审核发现的不合格项;再次是顾客对产品质量投诉。
2.5 对产品功能或性能的改进
主要包括高光电转换效率的激光器上市、高清晰度的智能液晶屏上市、新型高可靠性CPU上市和先进的集成电源控制模块上市等。
3 激光治疗设备常见设计开发更改导致的风险及其危害
以下列举了对激光治疗设备常见设计开发更改导致的风险及其危害的分析,具体见表1。
4 激光治疗设备设计开发中常见关键元器件的更改及应对控制措施
激光治疗设备的具体结构及其配件、附件因不同用途而各不相同,但基本包括电源系统、激光器、冷却系统、控制与防护系统和激光传输系统。如固体激光器由谐振腔、工作物质、泵浦源这3个关键组件组成,全反射镜和输出反射镜组成谐振腔,谐振腔一般需要机加工件来强化整个结构,并采用冷却水循环的方式降温,工作物质被固定在谐振腔中,泵浦源一般是高压电极。当泵浦源激励工作物质(释放高压时),工作物质受到激励释放光子,由于全反镜和输出反射镜的作用,激光会在谐振腔中反复震荡,直到达到输出阈值才会发射出激光。一般激光要经过传输耦合系统到达治疗部位,激光传输系统包括刀头、光纤、机械导光关节臂等几种类别。设计开发中常见关键元器件的更改及企业合理的应对控制措施见表2。
5 激光治疗仪注册后设计开发更改的常见识别方法
常见的识别注册产品设计开发更改的方法,包括以下3种。
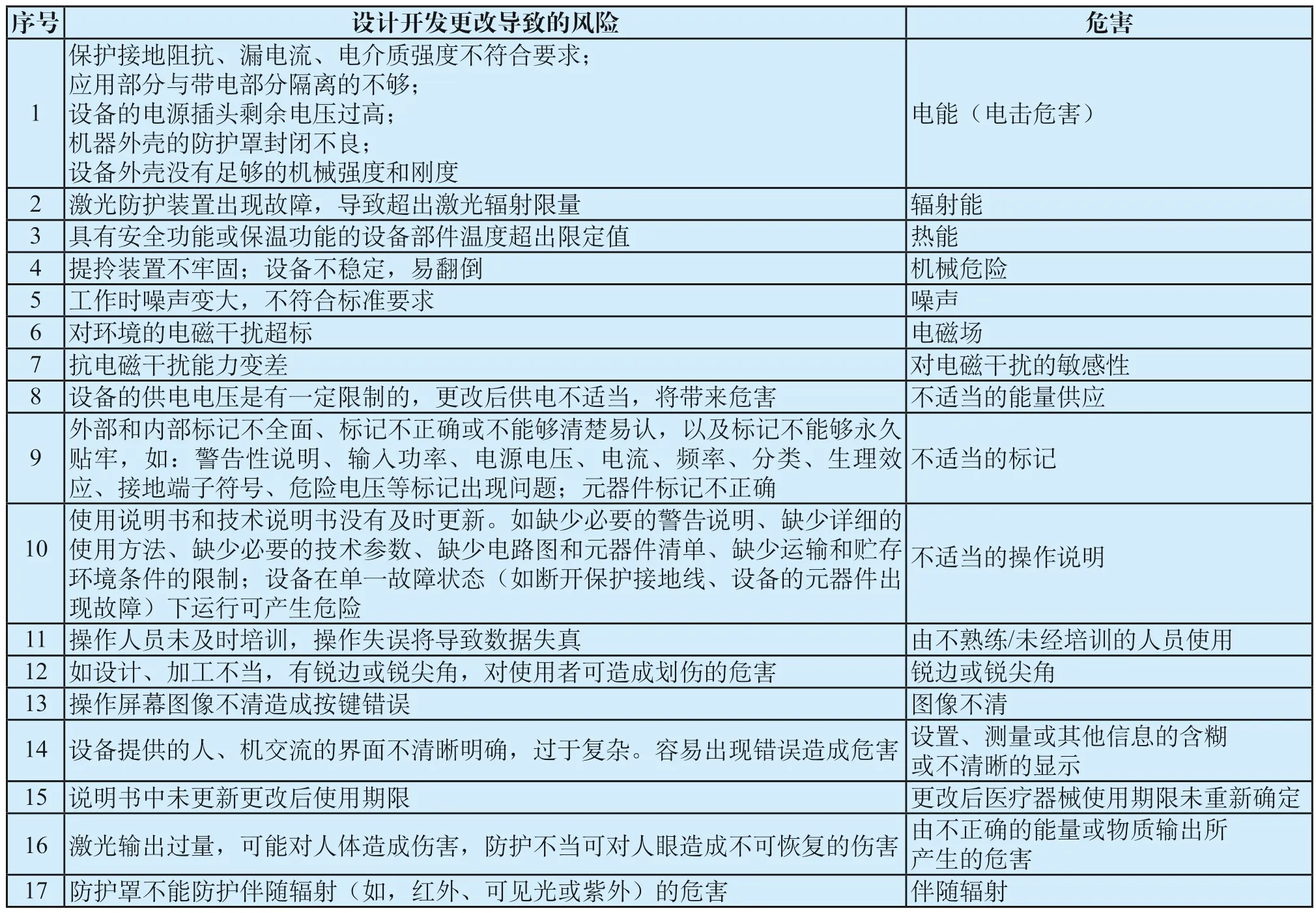
表1 常见设计开发更改导致的风险及其危害Tab.1 Risks and hazards caused by common design and development changes
5.1 注册检验报告相关信息与实物比对
注册检验过程中出现问题最多的项目是激光性能、通用电气安全(GB 9706.1[16])、电磁兼容(YY 0505—2012[17])。激光性能检验不合格导致设计开发更改的情况见表3,电气安全检验不合格导致设计开发更改的情况见表4,电磁兼容检验不合格导致设计开发更改的情况见表5。
部分医疗器械检验机构会在产品检验报告的检验项目中注明“*”,表示该项目是经过整改后合格的,从以上表格可以看出对应项目整改常需要加入或更换屏蔽壳、磁环、开关电源、变压器、电容、TVS管、压敏电阻、电感、电路板、滤波器、屏蔽线、铜箔说明书、标记等。
需要注意的是整改过程可能未全面考虑EMC与安规(GB 9706.1)之间的相互影响,导致已通过的项目在整改后测试不合格。如果EMC与安规报告未关联,不能表明两者测试使用的是同一台样机,企业未将注册检验过程中的整改内容引入到设计开发文档输出文档,也会导致上市的产品不符合标准要求而产生危害。EMC整改后与安规测试项目之间可能的相互影响见表6。
结合产品注册或委托检验报告中记录的关键元器件表、整机及局部照片等信息,与现场实物做对比,核查发生过哪些设计更改。有部分检验中心将产品在检验过程中进行过的整改以文件形式记录下来,交由企业留档,以供查阅。对于产品EMC与安规报告未关联的情况,也可通过比对安规检验报告、EMC检验报告和现场实物的方式判断是否为同一样机。

表2 常见关键元器件的更改及企业应对控制措施Tab.2 Change of common key components and control measures for enterprises
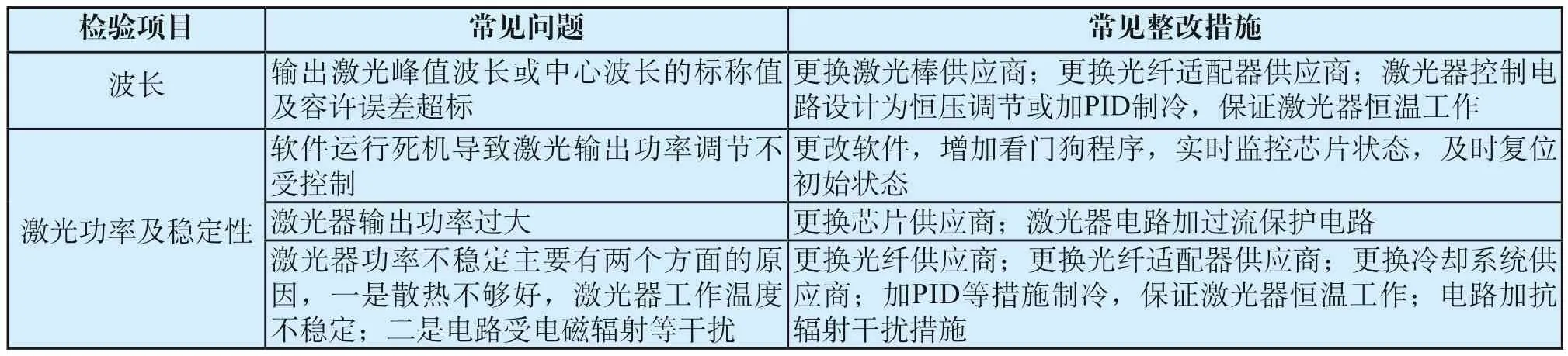
表3 激光性能检验不合格导致设计开发更改的情况Tab.3 Failure of laser performance inspection leads to change of design and development

表4 电气安全检验不合格导致设计开发更改的情况Tab.4 Failure of electrical safety inspection leads to change of design and development

表5 电磁兼容检验不合格导致设计开发更改的情况Tab.5 Failure of EMC inspection leads to change of design and development

表6 EMC整改与安规测试项目可能的相互影响Tab.6 Possible interaction between EMC rectification and safety test items
5.2 注册许可信息与实物比对
核查注册证、产品技术要求、说明书等注册许可事项中的信息与现场实物是否一致,所遇到的常见问题见表7。
5.3 设计开发文档与生产原料清单、仓库库存清单、采购凭证、生产工艺文件、检验记录、生产记录相互比对
设计开发文档中输出内容包含以下信息:采购信息,如原材料、包装材料、组件和部件技术要求;生产和服务所需的信息,包括产品图纸(包括零部件图纸)、工艺流程、作业指导书;产品使用说明书。
设计开发文档、生产原料清单、仓库库存清单、采购凭证、生产工艺文件、检验记录、生产记录中的原料信息应相互一致,抽查一批关键原料,检查其涉及的采购申请单、购货合同、购货发票或送货单、供应商检验报告、物料到货验收单、入库单、原料进货检验记录、领料单、生产记录等,核实关键原料的名称、供应商、型号、规格、照片等信息是否与设计开发输出以及注册检验报告内容一致。

表7 注册许可信息与实物比对的常见问题Tab.7 Common problems in comparison between registration license information and physical object
6 设计开发更改后处理措施
医疗器械设计和开发更改,特别是重要原材料、零部件或产品软件组件的更改,可能对产品安全有效性相关的其他质量特性产生较大影响。因此任何设计和开发更改,企业均需按设计和开发程序规定进行控制和管理。必要时,通过设计和开发策划、输入、输出控制,适当的评审、验证、确认及其每个阶段的后续措施,保证上市后的更改得到识别与控制。多项设计和开发更改同时进行,或更改可能导致产品安全性及有效性产生重大变化,企业应重新依次对与产品有关的更改要求、设计和开发更改的输入输出、原设计产品安全有效和己交付产品影响等进行系统的评审,开展风险分析并制定更改方案,批准后实施。更改后进行系统的验证和确认活动。如上述更改涉及注册证、技术要求、说明书的许可事项更改,还应按法规规定履行注册更改程序,确认产品是否再次进行型式检验,取得医疗器械更改注册后或完成相关法规规定义务后,将设计更改后的产品投放市场。同时企业应确认是否需要按规定对己交付产品实施召回。
