侧链含蒽可光交联聚芳醚酮的制备与表征*
2020-08-03田炎鑫何禹林
田炎鑫,张 辉,李 佳,刘 攀,何禹林,李 明,
(长春工业大学 化学与生命科学学院 吉林省碳纤维开发与应用重点实验室, 长春130012)
0 引 言
聚芳醚酮是一种具有较高的耐热稳定性、耐辐射性、耐腐蚀性、优良的力学性能和耐化学稳定性等特点的特种材料,在航空,航天,核能,电子信息,机械精密仪器和医疗器材上有着广泛的应用。聚芳醚酮其结构是由醚键和羰基交替连接刚性基团结构的线性聚合物[1-5]。传统的结晶型聚芳醚酮玻璃化转变温度较高,自身溶解性差,使其热塑加工条件苛刻,也难以通过溶液加工,从而限制聚芳醚酮在实际生产生活中的应用[6-7]。近几年来,为了满足实际生活中对聚芳醚酮需求的更高要求。为此,科学家通过分子设计合成出一系列改性聚芳醚酮来改良其加工性能,功能化聚芳醚酮是一种良好的途径[8-13]。其中可交联的聚芳醚酮引起了很多人的关注,交联前具有较低的玻璃态转化温度,容易进行热塑成型,使其具有易加工性。交联后使其形成网络聚合物,使其具有热固型材料的特征,来增强其热稳定性[14-19]。
在本文中,我们合成了一系列含蒽基团量不同的可光交联聚芳醚酮。该光交联是基于蒽环之间由365 nm波长的紫外灯引发的D-A环加成反应。该光交联反应不需要引入额外的光引发剂,避免了可能引起材料性能下降的因素[20-22]。该系列聚合物具有较低的玻璃态转化温度,可以在较低的温度下进行热塑成型。在加工成型后,通过低功率的紫外灯进行照射,引发蒽基团发生反应来光固化。使线性聚芳醚酮形成交联网状结构,其热稳定性和玻璃态转化温度提高。从而在不牺牲聚芳醚酮热稳定性的同时,改良了其加工性能。
1 实验部分
1.1 主要原料
4,4-双(4-羟苯基)戊酸(DPA),分析纯,98%,阿拉丁生化科技有限公司。4,4-二氟二苯甲酮,分析纯,99%,麦克林生化科技有限公司。4-二甲氮基吡啶(DMAP),99%,分析纯,阿拉丁生化科技有限公司。 N,N-二环己基碳二亚胺(DCC),分析纯,99%,阿拉丁生化科技有限公司。蒽甲醇,分析纯,99%,麦克林生化科技有限公司。N-甲基吡咯烷酮(NMP),分析纯,天津北联公司。 无水碳酸钾,分析纯,99%,天津光复科技公司。浓盐酸,分析纯,北京化工厂。四氢呋喃,分析纯,天津富宇公司。无水四氢呋喃将四氢呋喃通过钠丝精馏自制而得。甲醇,分析纯,北京化工厂。 乙醇,分析纯,北京化工厂。
1.2 主要仪器及测试
1H NMR和13C NMR谱图分别是由Bruker III HD 400光谱仪(400 MHz)和Bruker AVANCE (500 MHz)光谱仪中测得的。
紫外-可见光光谱是在Agilent Cary 5000光谱仪中测得的。
差式扫描量热法(DSC),在NETZSCH4标准模式下在氮气的保护下扫描速率为10 ℃·min-1测得的。
分解温度是通过Metter Toledo TGA 2热重量分析仪(TGA)在氮气的保护并且升温速率是10 ℃·min-1的条件下测得的。
红外图谱由Nicolet/iS 50傅立叶变换红外光谱仪(FT-IR)测得的。
聚合物分子量由LC98IIRI凝胶色谱仪(GPC)测得的。
1.3 聚合物P0,P1,P3和P5的合成
将单体双酚酸(11.57 g, 40 mmol)和4,4-二氟二苯甲酮(8.72 g, 40 mmol)在氮气下溶于NMP (60 mL)中。在该反应中加入K2CO3(8.28 g, 60 mmol)作为成盐剂。反应系统与机械搅拌混合。加入甲苯(40毫升)作为共沸剂,在140 ℃下脱除水4 h。然后将反应温度升高至170 ℃,持续6 h。将反应溶液倒入盐酸水溶液中(酸性水溶液pH控制在1~2之间),过滤后用水冲洗数次。然后将聚合物重新溶解在四氢呋喃中。采用真空吸滤装置和砂芯过滤装置对聚合物溶液进行过滤。去掉不溶杂质,将纯化后的聚合物溶液滴入甲醇中。聚合物溶液过滤后,在100℃的真空干燥箱中干燥过夜并收集得到聚合物P0,所得产物的产率为75.6%。1H NMR (400 MHz, DMSO) δ 7.75 (s, 4H), 7.27 (s, 4H), 7.07 (s, 8H), 2.36 (s, 2H), 2.01 (s, 2H), 1.61 (s,3H)。
将P0 (4.66 g, 10 mmol)和9-蒽甲醇(0.2082 g, 1 mmol)分别放入圆底烧瓶中,溶解于干燥的THF (25 mL)中。加入DMAP (0.1 22 mg 1 mmol)时,滴加DCC溶液(5 mL THF溶剂2.472 g)。反应在室温下氮气下搅拌8h。然后,将乙醇加入到反应溶液中。反应体系仍然被搅拌了2 d。最后,将反应溶液在0 ℃下冷藏2 h进行过滤。滤液注入甲醇。过滤得到聚合物P1, 60 ℃真空干燥过夜,其产率为73.5%。1H NMR (400 MHz, DMSO) δ 8.57 (s, 0.08H), 8.33 (s, 0.29H), 8.10 (s, 0.23H), 7.75 (s, 4.66H), 7.56 (s, 0.41H), 7.26 (s, 4.3H), 7.07 (s, 8.2H), 6.09 (s, 0.3H), 3.53 (s, 1.9H), 2.40 (s, 2.13H), 2.12 (s, 2.00H), 1.60 (s, 3.5H), 1.24 (s, 2.6H)。
聚合物P3的合成方法和P1相似,只是投料为P0 (4.66 g, 10 mmol)和9-蒽甲醇(0.6248 g, 3 mmol)。反应1d后加入微量乙醇反应掉多余的羟基。反应3d后沉入甲醇并过滤,所得产物的产率为74.5%。1H NMR (400 MHz, DMSO) δ 8.63 (s, 0.25H), 8.31 (s, 0.45H), 8.11 (s, 0 .65H), 7.74 (s, 4.36H), 7.54 (s, 0.98H), 7.25 (s, 4.36H), 7.06 (s, 8.2H), 6.09 (s, 0.3 H), 3.53 (s, 1.86H), 2.39 (s, 2.3H), 2.10 (s, 2H), 1.60 (s, 3.2H), 0.99 (s, 1.32H)。
P5的合成方法和P1相似,只是投料为P0 (4.66 g, 10 mmol)和9-蒽甲醇(1.041g, 5mmol)。反应2 d后加入微量乙醇反应掉多余的羟基。反应2d后沉入甲醇过滤,所得产物的产率为66.7%。1H NMR (400 MHz, DMSO) δ 8.67 (s, 0.52H), 8.34 (s, 1.07H), 8.11 (s, 1.3H), 7.76 (s, 4.2H), 7.53 (s, 2.3H), 7.28 (s,4.3H), 7.08 (s, 8.07H), 6.11 (s, 1H), 3.56 (s, 1.06H), 2.40 (s, 2.18H), 2.12 (s, 2H), 1.59 (s, 3.07H), 1.25 (s, 1.53H)。
2 结果与讨论
2.1 聚合物P1,P3和P5的合成与表征
合成光交联聚合物聚芳醚酮的合成路线如图1所示。首先以双酚酸和4,4-二氟二苯酮为原料,NMP为溶剂,合成侧链含羧基的聚芳醚酮预聚物,其分子量Mn=9 776 Da;Mw=11 427 Da。然后,通过酯化反应将蒽甲醇接枝到聚芳醚酮骨架上。合成出一系列含蒽量不同的可光交联聚芳醚酮。我们通过分析FT-IR和1HNMR图谱来证明成功制备蒽含量分别为10%、30%和50%的聚芳醚酮P1、P3和P5。图2为合成聚合物P0的红外光谱。缩聚反应结束后得到的产物中,在3 442 cm-1处的酚羟基特征峰已经消失,所得产物在1 709-1处出现羧基(C=O)的羰基伸缩振动峰,在1 648 cm-1处分别出现了属于酮基的羰基(C=O)的伸缩振动峰,表明酚羟基与二氟二苯甲酮亲核取代反应的成功进行。
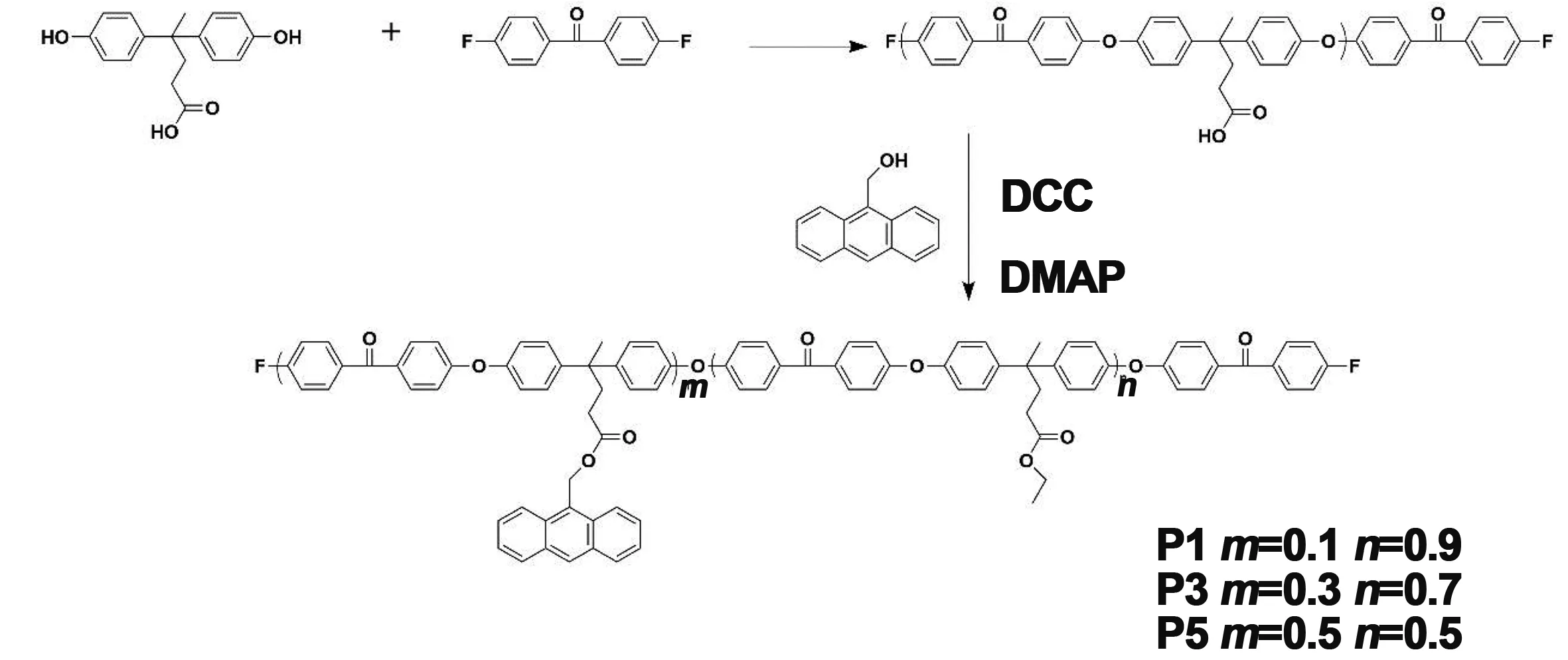
图1 光交联聚芳醚酮的合成路线Fig 1 Synthesis route of the photo-crosslinking poly(aryl ether ketone)
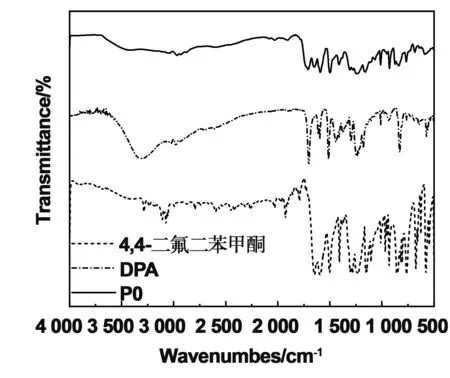
图2 前体聚合物P0的红外图谱Fig 2 FI-IR spectra of precursor polymer P0
图3为合成P5的FT-IR图谱,与聚合物P0相比,产物P5的羟基(-OH)的特征吸收带在3 457 cm-1处几乎消失,羰基(C=O)伸缩振动峰从1 709 cm-1移至1 731 cm-1,这是由于羧基反应变成酯键。表明酯化反应已经成功的将光交联基团蒽接入聚合物侧链。图4为P0,P1,P3和P5的1HNMR波谱图,由于蒽环的特征氢谱的强度相对于聚合物来说比较弱,本文选取化学位移(6~9)×10-6的核磁谱图进行放大分析。δ=7.75×10-6,7.27×10-6,7.07×10-6的吸收峰是属于聚芳醚酮高分子链中苯环的氢原子,而化学位移位于δ=8.61,8.31,8.11和7.54 ppm的氢谱对应的是蒽环的氢原子,也表明蒽基团的成功接入。我们通过分析计算图4中P1,P3和P5蒽的氢原子个数来确定蒽的在聚芳醚酮中的接入量。理论上蒽的接入量为100%时,在核磁谱图中蒽基团对应的质子数分别是1,2,2和4。其中聚芳醚酮P1蒽基团对应的质子数分别是0.08, 0.29, 0.23和0.41。实际蒽基的接入率在10%左右;聚芳醚酮P3蒽基团对应的质子数分别是0.25, 0.45, 0.60和0.98。实际蒽基的接入率在25%左右;聚芳醚酮P5蒽基团对应的质子数分别是0.52, 1.07, 1.3和2.3。实际蒽基的接入率在51%左右。综上所述,我们合成了一系列蒽含量不同的聚芳醚酮。
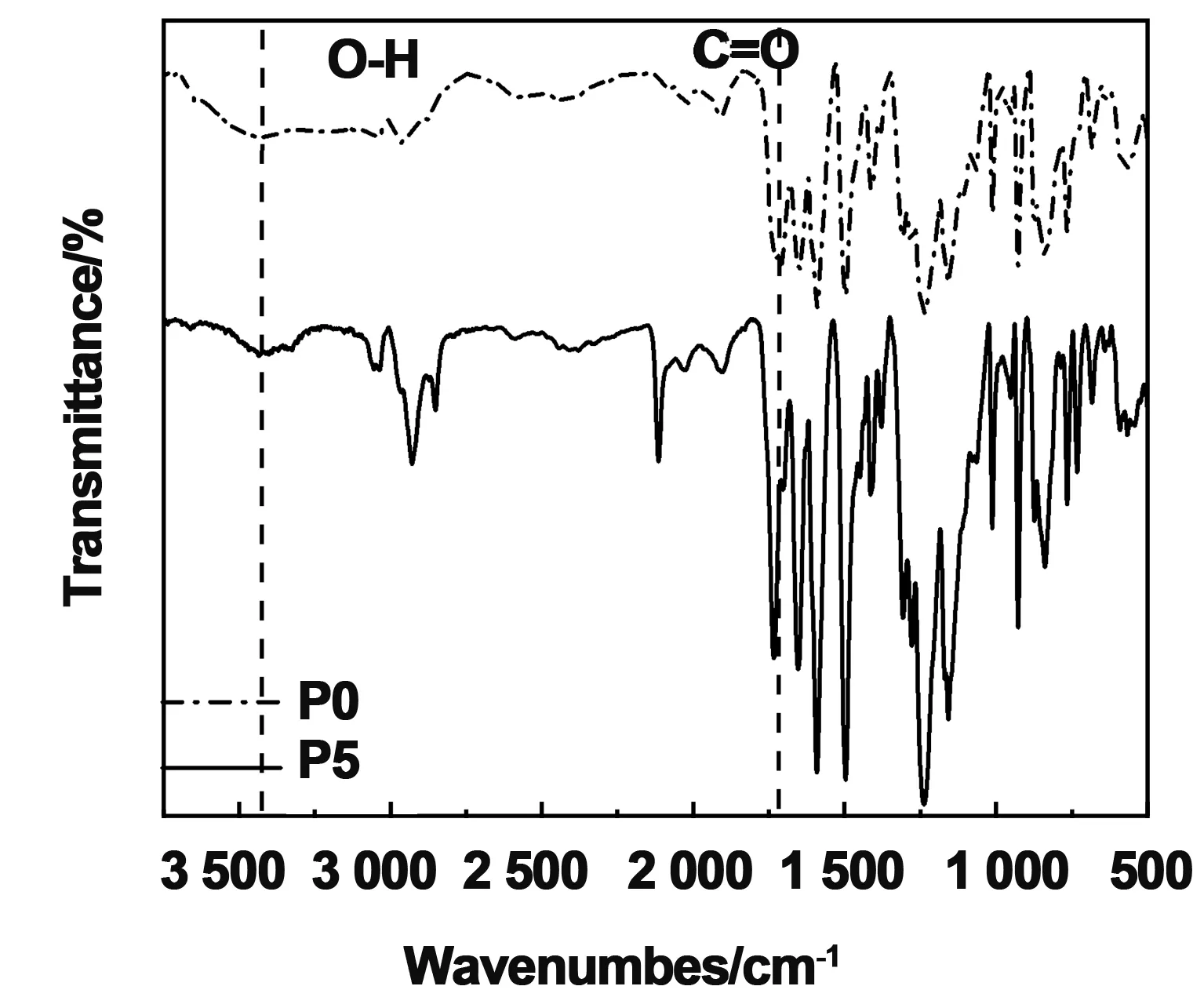
图3 聚合物P5的红外图谱Fig 3 FI-IR spectra of polymer P5
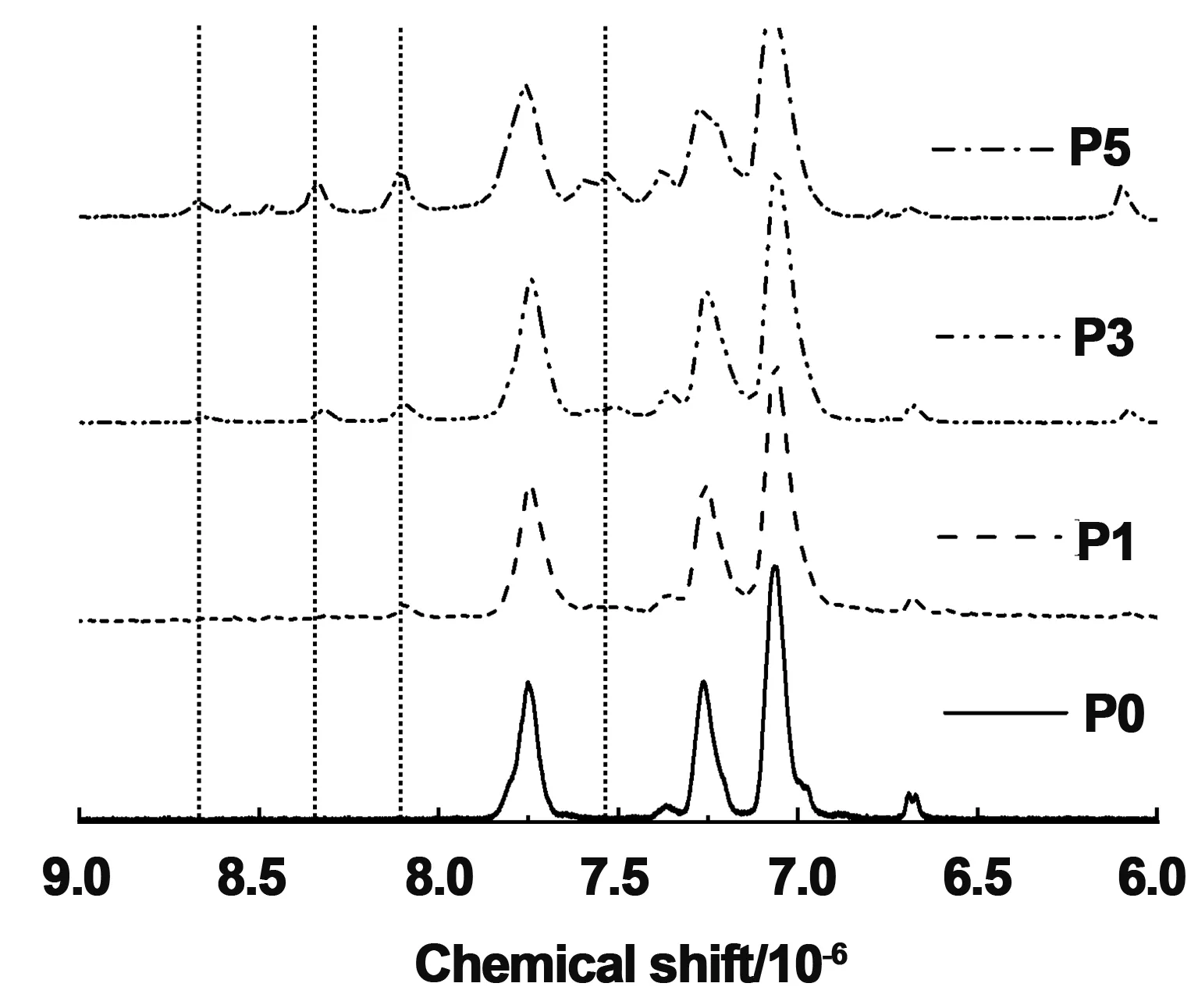
图4 聚合物P0,P1,P3和聚合物P5的核磁氢谱Fig 4 1H NMR spectra of polymer P0, P1, P3 and P5
2.2 光交联过程的研究
该系列聚芳醚酮是通过波长为365 nm的紫外光来引发光交联,并通过紫外可见吸收光谱图监测蒽基团交联反应进程。首先将一系列聚芳醚酮溶解在环戊酮中配制成浓度是300 mg/mL的聚合物。再通过旋涂制备得到2~3 μm的聚合物薄膜。使用手提低功率紫外灯照射薄膜,完成光交联过程。蒽基的吸收峰是位于280~390 nm之间呈多重峰。如图5(a)和图5(b)所示,在紫外光照10 min后,波长在280~390 nm处的蒽特征吸收峰吸光度大幅度减弱。在光照20 min后,波长在280~390 nm处的蒽特征吸收峰不在减弱。表明在光照20 min后,蒽基团的交联反应基本完成。
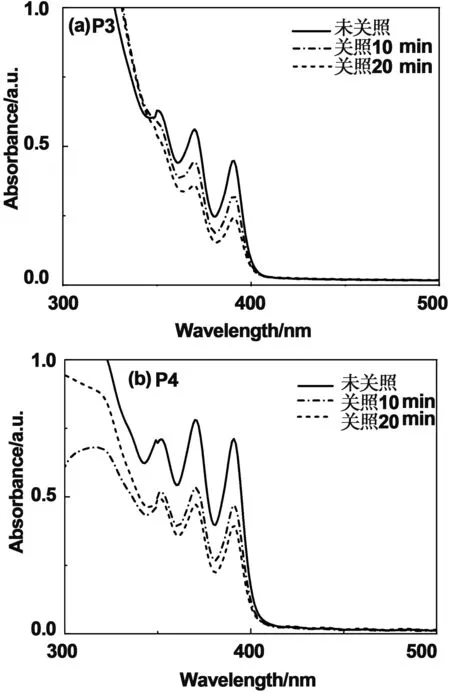
图5 P3和P5在不同紫外光照时间下的紫外图谱Fig 5 UV-vis absorption spectra of P3 and P5 in different ultraviolet light (λ=365 nm) illumination time
2.3 交联前后的热力学表征
我们采用扫描差示量热法(DSC)和热重分析法(TGA)表征交联聚芳醚酮聚合物的的热稳定性。P3和P5的DSC曲线图如图6(a)和 (b)所示,其中聚合物P3和P5在未交联前的玻璃化转变温度(Tg)分别为116和118 ℃,这表明可以在较低的加工温度下热塑成型,交联后的P3和P5都在180℃以下没有明显的玻璃态转化温度,表明有良好的耐热性。而TGA数据如图7所示,交联前P3和P5的热分解温度(Td5%)分别是234和252 ℃。当该材料成型交联后,与交联前相比,交联后的P3的热失重5%(质量分数)的温度(Td5%)提高了65 ℃,而P5的5%的(Td5%)提高了91 ℃。证明了交联后其热稳定性大幅提高。所有的数据分别总结于表1中。通过使用DSC和TGA测量光交联前后聚合物的热力学性能研究,表明蒽修饰的聚芳醚酮具有较低的热加工温度。而在交联后,他们的热稳定性明显提高,在优化聚芳醚酮加工性能的同时也保证了其具有一定的热稳定性。而且所有聚合物在未交联前。均在一系列有机溶剂如四氢呋喃,N,N-二甲基甲酰胺,二甲基亚砜和环戊酮中均表现出良好的溶解性。该系列材料也可以通过溶液法来加工成型。
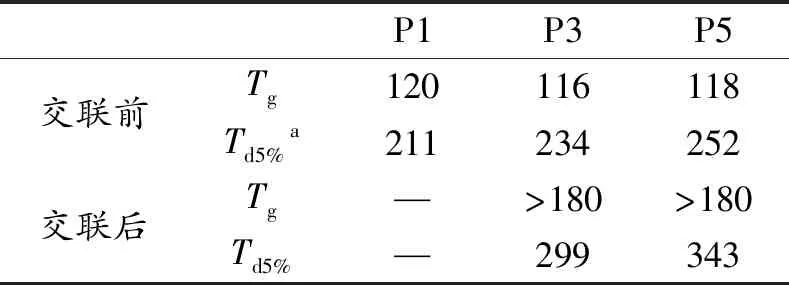
表1 交联前和交联后聚合物的热力学性能
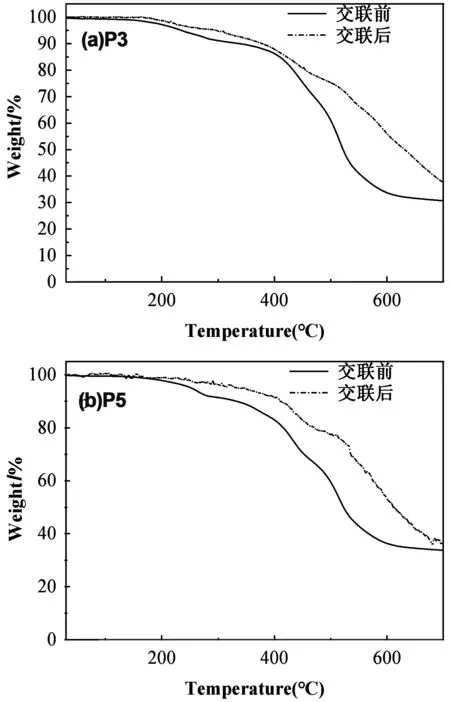
图7 P3和P5交联前后的TGA曲线Fig 7 TGA curves of P3 and P5 before and after cross-linking
由于测试DSC的环境因素,实验室所有的DSC曲线都会在100 ℃出一个小峰。
3 结 论
本文设计合成了一系列含有可光交联基团蒽的聚芳醚酮。采用红外光谱、核磁氢谱表征其结构,扫描差示量热法(DSC)分析结果表明蒽接枝的聚芳醚酮玻璃态转化温度在116~120 ℃之间,较低的玻璃态转化温度赋予了他们良好的可加工性和加工条件。采用紫外可见光谱法确定最佳光交联条件,DSC和热失重分析(TGA)结果表明聚芳醚酮的热稳定性明显提高,交联后热失重5%(质量分数)温度可达343 ℃。系列研究结果表明蒽功能化的聚芳醚酮在交联前具有热塑型材料的良好加工性能,在交联后有较高的耐热性和热稳定性。有望解决聚芳醚酮难以加工的问题,拓宽了聚芳醚酮的应用前景,为开发新型特种塑料提供了一个具有前景的展望。
