Mitochondrial translation defects and human disease
2020-07-30BrynWebbGeorgeDiazPankajPrasun
Bryn D.Webb, George A.Diaz, Pankaj Prasun
Department of Genetics & Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Abstract
Keywords: Mitochondria, translation defect, tRNA, aminoacyl-tRNA synthetase, rRNA, ribosomal protein, mitochondrial disease, mtDNA
INTRODUCTION
Mitochondria are double-membrane bound organelles found in most eukaryotic organisms with the important function of generating cellular energy via oxidative phosphorylation, but which also function in cellular signaling, cellular differentiation, cell death, and cell cycle regulation.Mitochondria are estimated to be comprised of approximately 1100 proteins and are unique organelles in that they have their own genome and ribosomes that carry out protein synthesis inside the mitochondria[1].The mitochondrial genome, which is housed in the mitochondrial matrix, encodes 37 genes: 13 which encode protein subunits of respiratory Complexes I, III, IV, and V; 22 which encode mitochondrial tRNAs; and 2 which encode mitochondrial rRNAs.By far the majority of mitochondrial proteins are produced on cytosolic ribosomes and are transported to the mitochondria as precursors via the translocase of the mitochondrial outer membrane (TOM complex), the presequence translocase (TIM23 complex), and presequence-translocase-associated motor located at the inner mitochondrial membrane[2].
Oxidative phosphorylation and generation of cellular ATP requires coordinated biogenesis and assembly of respiratory chain complexes at the inner mitochondrial membrane.Electrons are transferred along the respiratory chain complexes from the reducing equivalents NADH and FADH2 to oxygen to produce water and generate a proton gradient across the inner membrane.This proton gradient enables ATP synthase to generate ATP from ADP and phosphate.In humans, five multi-subunit protein complexes compose the respiratory chain and oxidative phosphorylation system: NADH dehydrogenase (Complex I); succinate dehydrogenase (Complex II); coenzyme Q: cytochrome c-oxidoreductase (Complex III); cytochrome c oxidase (Complex IV); and ATP synthase (Complex V).Complex II is composed of proteins encoded entirely by the nuclear genome, whereas the remaining complexes have protein subunit components encoded by both nuclear and mitochondrial genomes.Additionally, complex assembly is a highly coordinated process involving a number of assembly factors, as well as coordination of nuclear and mitochondrial genes.Defects in mitochondrial translation processes may result in impaired activities of these complexes, resulting in deficient aerobic energy metabolism and clinical disease in humans[3].
Structural studies have established that many mitochondrial ribosome proteins have eubacterial orthologs, but there also exist additional proteins without such orthologs.Mitoriboproteins have traditionally been named by a MRPS (Mitochondrial Ribosomal Protein Small subunit)/MRPL (Mitochondrial Ribosomal Protein Large subunit) nomenclature[5].Recently, a new naming convention has been proposed based on functional/structural relationships of mitoribosomal proteins across species in order to reduce ambiguity arising from non-orthologous proteins from different species being assigned similar names[7].
Mitochondrial translation defects resulting in human disease may have varying organ involvement, varying age of onset, and varying modes of inheritance.This specific class of mitochondrial disease may be caused by the following mechanisms: mitochondrial tRNA mutations, mitochondrial aminoacyl-tRNA synthetase mutations, mitochondrial rRNA mutations, and mitochondrial ribosomal protein mutations.Additional mechanisms of abnormal mitochondrial translation exist, including impaired translation secondary to mtDNA depletion and defects in mitochondrial RNA synthesis, modification, and degradation, which are beyond the scope of this article but have been recently reviewed[8,9].

Table 1.Clinical phenotypes of mt-tRNA disorders
MITOCHONDRIAL tRNA MUTATIONS
All 22 mt-tRNAs are encoded by the mitochondrial genome, and the primary function of mt-tRNAs is to deliver amino acids to the nascent polypeptide chain during mitochondrial protein translation.Mitochondrial tRNAs are truncated when compared to their canonical cytosolic tRNA counterparts, and, in some cases, such as in tRNASer(AGY), one arm of the classic cloverleaf secondary structure of tRNA is lost[10].
The first report of a mt-tRNA mutation causing human disease was published in 1990 when Kobayashiet al.[11]revealed that a mutation in the mitochondrial tRNALeugene (MTTL1) was causative of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS)[11,12].Since then, over 300 mutations in mt-tRNA genes have been identified to cause human disease [Table 1].Most of these mutations prevent tRNA aminoacylation.mt-tRNA mutations have been identified in various structural locations including in the anti-codon wobble position, anti-codon stem, acceptor stem, DHU stem, TYC stem, and the variable loop[12].Disorders associated with mitochondrial t-RNA mutations are summarized in Table 1.
Interestingly, different point mutations in the same mt-tRNA molecule can result in different human diseases.For example, the point mutation m.14709T>C inMTTE(gene that encodes the mitochondrial tRNAGlu) can result in the phenotype of maternally inherited diabetes and deafness, whereas the point mutations m.14674T>G or m.14674T>C inMTTEcan result in infantile transient mitochondrial myopathy.
Nearly all mitochondrial disease resulting from mt-tRNA mutations display maternal inheritance as mitochondrial DNA is inherited from the mother.However, few instances of paternal inheritance have been reported as well[13,14].Cells that carry a homogeneous population of the mitochondrial genome, either wildtype or mutant, are termed homoplasmic.Cells that carry two or more populations of the mitochondrial genome are termed heteroplasmic.With mitochondrial disease due to maternal inheritance, clinical disease severity often correlates with mutation load in affected tissues.
The two most well-known mitochondrial diseases associated with mt-tRNA mutations are MELAS and myoclonic epilepsy with ragged red fibers (MERRF).In approximately 80% of MELAS patients, the causative mutation is the m.3243A>G pathogenic variant inMTTL1(mt-tRNALeu).Most patients with MELAS develop symptoms between ages 2 and 40 years old, and these symptoms include stroke-like episodes, encephalopathy with seizures and/or dementia, muscle weakness, exercise intolerance, headaches, vomiting, hearing impairment, peripheral neuropathy, learning disability, and short stature.Treatment for MELAS is supportive and includes treatment with a mitochondrial cocktail.Intravenous arginine is recommended during acute stroke-like episodes, and arginine should be given orally for prophylaxis after a patient has had a first stroke-like episode[15].
The most common mutation causing MERRF in more than 80% of affected patients is the m.8344A>G mutation inMTTK(mt-tRNALys).Onset of MERRF is usually in childhood and the first symptom is often myoclonus.Other common symptoms and findings are epilepsy, ataxia, weakness, dementia, hearing loss, short stature, optic atrophy, and cardiomyopathy with Wolff-Parkinson-White syndrome.Treatment is also supportive with antiepileptic medications to treat seizures, mitochondrial cocktail, and physical therapy[16].
通过前期招标,管理企业先是获得项目管理的代理权,并与市政相关部门及项目委托单位签订《项目前期工作合同》。依据国家政府及各地方政府相关的法律条例,管理企业应组织项目设计招标会,选定中标企业完成项目的勘察及设计工作。遵照合同,工程项目开始进入前期工作阶段,其内容主要是现场勘察、初步设计及成本预估等。初步设计审核、预算申报、相关建设手续的审批等诸多事务需要有序进行,协助筹集资金、加强资金管理、申报建设许可证、图纸审核与审批等,均是建设项目管理需要注意的节奏时点,这些环节是项目运用节奏管理需注意的要点,掌握好各环节中的节奏,可避免流程出现混乱,影响项目的有序进行。
MITOCHONDRIAL AMINOACYL-tRNA SYNTHETASE DISORDERS
Mitochondrial aminoacyl-tRNA synthetases (mt-ARSs) are essential for protein synthesis in the mitochondria and generation of oxidative phosphorylation (OXPHOS) system components.mt-ARS proteins are nuclear-encoded and function to charge mitochondrial tRNA molecules, which are mitochondrial-encoded, with their cognate amino acids.While mt-ARS proteins vary in size and oligomeric state (from monomer to tetramer), all contain a catalytic domain and a tRNA anticodon-binding domain[17].mt-ARS genes are named with anARS2nomenclature (for example,MARS2for methionine tRNA synthetase, mitochondrial).For the amino acids glycine and lysine, a separate mt-ARS gene does not exist, andGARSandKARS, respectively, function as the aminoacyl-tRNA synthetase in both the cytosol and the mitochondria.Additionally, an mt-ARS has not been identified for glutamine (Q), and Q-tRNA is believed to be formed by postconjugation modification of glutamate[18].
The first Mendelian disease reported to be caused by mt-ARS mutations was leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation (MIM #611105) due to autosomal recessive pathogenic variants inDARS2, which was reported in 2007[19].Since then, pathogenic variants in all known mt-ARSs have been identified with the majority being identified by whole exome sequencing studies, and the associated conditions represent a new class of Mendelian disorders [Table 2].All mt-ARS disorders exhibit autosomal recessive inheritance and most often patients are compound heterozygotes[17].These Mendelian disorders are extremely rare as deleterious mutations in mt-ARS genes leading to absent mt-ARS function are expected to be lethal.Therefore, patients most often have at least one allele with a mild mutation leading to some residual mt-ARS gene function.
Interestingly, although pathogenic variants in all mt-ARSs are expected to result in disruption of protein synthesis of OXPHOS system components via impairment of mitochondrial translation, the identified mt-ARS disorders each display strikingly specific clinical phenotypes with specific tissue involvement [Table 2][17,20,21].Most frequently, mt-ARS disorders display central nervous system involvement, but additional organ systems are specifically involved in certain disorders, such as ovaries in the case ofHARS2andLARS2or kidney in the case ofSARS2[Table 2][22-24].Additionally, age of onset is highly variable for the various mt-ARS disorders.The molecular mechanisms behind this selective tissue involvement and disease phenotype for specific mt-ARS disorders are currently poorly understood.

Table 2.Clinical phenotypes of mt-ARS disorders
A wide variety of neurological symptoms is also seen with mt-ARS disorders.Leukoencephalopathy may be seen withAARS2,DARS2,EARS2,NARS2,PARS2, andWARS2disorders.Epilepsy may be seen withCARS2,EARS2,FARS2,NARS2,PARS2,RARS2,TARS2,VARS2, andWARS2disorders.Peripheral neuropathy is seen withIARS2disorder.Sensorineural hearing loss may be seen withHARS2,IARS2,LARS2,MARS2,NARS2, andPARS2disorders[18].
Pathogenic mutations inGARS,which functions in both the cytosol and mitochondria, may cause autosomal dominant Charcot-Marie-Tooth disease, type 2D (MIM #601472) or autosomal dominant neuropathy, distal hereditary motor, type VA (MIM #600794)[25].Additionally, a few cases ofGARSvariants causing autosomal recessive disease have been reported leading to cardiomyopathy or complex neurological phenotypes [Table 2].Pathogenic mutations inKARSmay cause autosomal recessive Charcot-Marie-Tooth disease, recessive intermediate B (MIM #613641) or deafness, autosomal recessive 89 (MIM #613916)[26,27].Interestingly, Ruzzenenteet al.[28]recently reported a patient with compound heterozygousKARSvariants leading to impaired mitochondrial translation, but intact cytosolic translation.This patient had symptoms of sensorineural deafness, developmental delay, hypotonia, and lactic acidosis[28].Additional case reports have described additional various phenotypes for patients with pathogenicKARSmutations including optic neuropathy, progressive leukoencephalopathy, and cardiomyopathy, among others[29-31].
Failure of charging of glutaminyl mt-tRNA (mt-tRNAGln) has also been identified to cause disease.The GatCAB aminoacyl-tRNA amidotransferase complex provides this function and is composed of three subunits: GATA encoded byQRSL1, GATB encoded byGATB, and GATC encoded byGATC.Patients with defects in glutaminyl mt-tRNA charging present in infancy with lethal cardiomyopathy and lactic acidosis.Pathogenic variants have been identified inQRSL1,GATB, andGATC, and all cause autosomal recessive disease[32,33].
In addition to mt-ARS genes functioning in mitochondrial translation, there is growing evidence that mt-ARS proteins have potential non-canonical roles in immune regulation, inflammation, and neuronal differentiation[34].Further work is in progress to further explore the many roles of mt-ARS genes.
MITOCHONDRIAL rRNA MUTATIONS
Mitochondrial 55S ribosomes are composed of two subunits.The small 28S subunit (mtSSU) functions to catalyze the peptidyl-transferase reaction and the large 39S subunit (mtLSU) functions in mt-mRNA binding and decoding[8].The 28S and 39S mitochondrial ribosome subunits are composed of 12S mt-rRNA (mtSSU) and 16S mt-rRNA (mtLSU) and ribosomal proteins.Both mt-rRNAs are processed from the polycistronic heavy strand transcript, which also encodes tRNAPheand tRNAVal.Following release of the mature mt-rRNAs by endonucleolytic cleavage, assembly of the functional mitoribosome proceeds via a complex process involving maturation and processing of mt-rRNAs and association with ribosomal proteins[35].In addition to the 16S mt-rRNA, the large subunit of mammalian ribosomes also include tRNAPheor tRNAVal[36,37].
The geneMTRNR1encodes the mitochondrial 12S ribosomal RNA, and the geneMTRNR2encodes the mitochondrial 16S ribosomal RNA.Mutations inMTRNR1are associated with hearing impairment with or without aminoglycoside exposure.TheMTRNR1mutations m.1555A>G[38]and m.1494C>T[39]have been described as a cause of maternally inherited deafness in numerous case reports but the phenotype is variable and not completely penetrant.The identification of a pedigree in which deafness manifested when the m.1555A>G variant was co-inherited with a loss-of-functionSSBP1variant suggests that SSBP1 may be a phenotypic modifier of m.1555A>G-associated deafness[40].Additional examples of complex phenotypes involving m.1555A>G include in a pedigree in which the hearing loss co-segregated with familial dilated cardiomyopathy due to mutations inMT-ATP6[41].Recently, expansion of theMTRNR1poly-cytidine tract at m.961 has been reported to be associated with non-ophthalmologic manifestations (intellectual disability, epilepsy, and migraine) in a kindred also segregating Leber’s hereditary optic neuropathy due to m.3460G>A, although this association is not statistically validated[42].In contrast to the numerous reports of human disease-associated variants inMTRNR1, only a single variant inMTRNR2, m.2336C>T, has been identified as a cause of hypertrophic cardiomyopathy in humans[43].
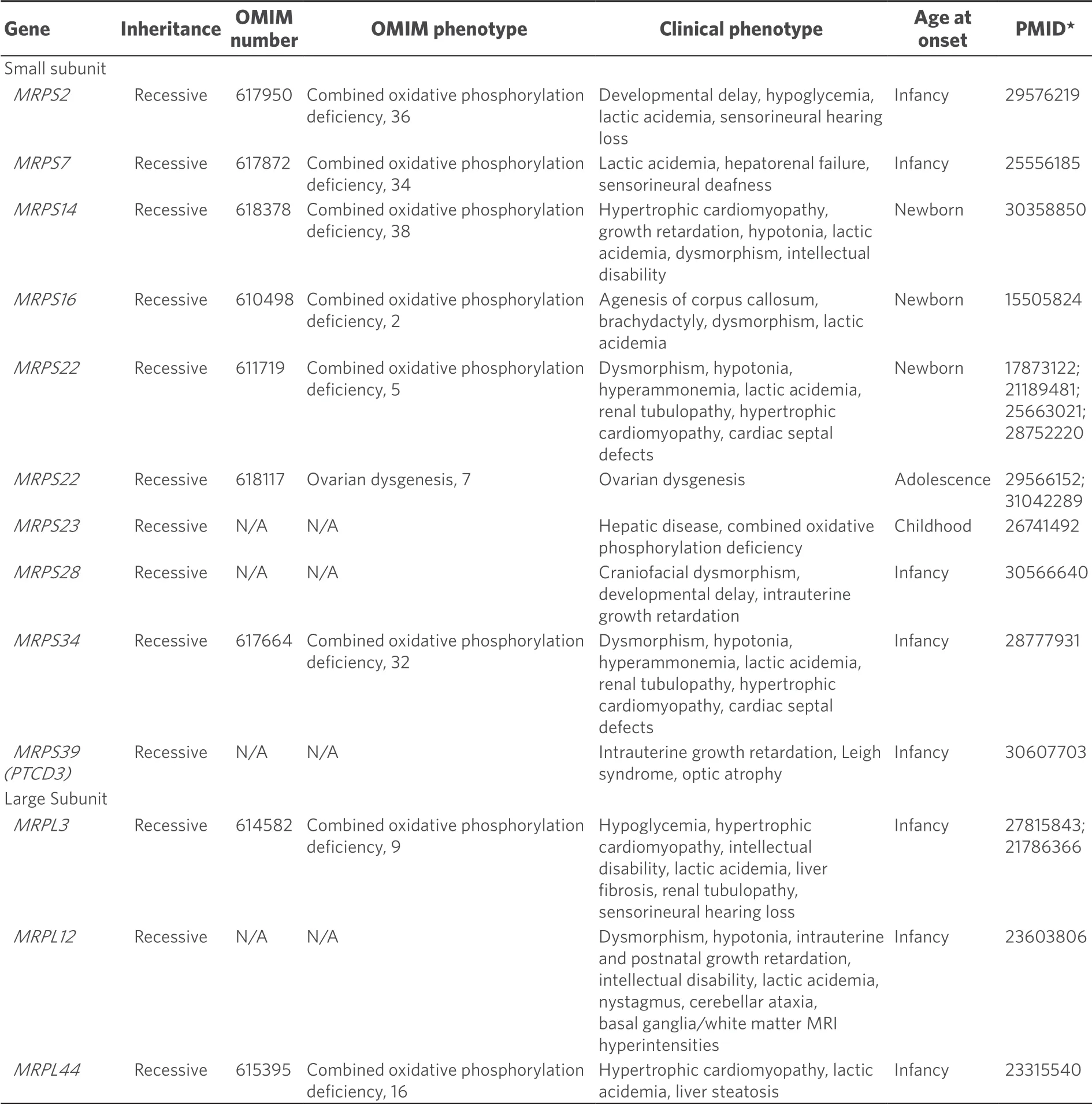
Table 3.Clinical phenotypes of mitochondrial ribosomal protein disorders
MITOCHONDRIAL RIBOSOMAL PROTEIN DISORDERS
Mammalian mitoribosomes are composed of rRNA and mitochondrial ribosomal protein components.In humans, 30 mitochondrial ribosomal small subunit proteins (MRPSs) assemble with the 12S mt-rRNA to form the small mitoribosomal 28S subunit.Similarly, 50 mitochondrial ribosomal large subunit proteins (MRPLs) assemble with the 16S mt-rRNA along with tRNA to form the large mitoribosomal 39S subunit[44].MRPS and MRPL proteins are all encoded by the nuclear genome.
At present, nineMRPSgenes and threeMRPLgenes have been identified to cause mitochondrial disease in humans [Table 3].All are inherited in an autosomal recessive fashion.Mutations in mitochondrial ribosomal protein genes may destabilize the mitoribosomal subunits impacting translation, as has been shown via proteomic analysis withMRPS34disorder[44].Despite the presumptive shared pathogenesis of destabilizing either the large or small mitoribosomal subunits, the clinical phenotypes associated with mutations in genes encoding mitoribosomal structural proteins are surprisingly diverse [Table 3].Most of these disorders present early in life, although missense mutations inMRPS22can present with ovarian failure in adolescent females[45].Neurological deficits have been observed in the majority of patients with this subset of disorders but additional associated clinical phenotypes include hepatopathy, renal dysfunction, deafness, myopathy, and craniofacial or cardiac phenotypes.The neurological features may be variable and range from structural lesions such as agenesis of the corpus callosum to classical Leigh syndrome or functional deficits without apparent structural lesions.Most mitochondrial ribosomal protein subunit disorders cause severe disease often with multi-organ involvement and early death.In the future, additional mitochondrial ribosomal protein disorders are highly likely to be identified via whole exome sequencing of patients with suspected mitochondrial disease.
CONCLUSION
Defects in mitochondrial translation may result in a vast array of clinical disease.Disease mechanisms include, but are not limited to, mitochondrial tRNA mutations, mitochondrial aminoacyl-tRNA synthetase mutations, mitochondrial rRNA mutations, and mitochondrial ribosomal protein mutations.Understanding disease biology of these mitochondrial translation defects is a necessary predecessor to developing effective treatment for these disorders.More research is necessary to further understand this emerging class of mitochondrial disease.
DECLARATIONS
Authors’ contributions
Reviewed literature, wrote manuscript, and edited manuscript: Webb BD, Diaz GA, Prasun P
Availability of data and materials
Not applicable.
Financial support and sponsorship
Dr.Bryn D.Webb receives support from National Institutes of Health National Institute of Child Health and Human Development (K08HD086827).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2020.
猜你喜欢
杂志排行

Journal of Translational Genetics and Genomics的其它文章
- Intellectual disability, the long way from genes to biological mechanisms
- Spectrum of MECP2 mutations in Indian females with Rett Syndrome - a large cohort study
- The North American mitochondrial disease registry
- Role of transfer RNA modification and aminoacylation in the etiology of congenital intellectual disability
- Cryogenic electron paramagnetic resonance spectroscopy of flash-frozen tissue for characterization of mitochondrial disease
- Redefining infantile-onset multisystem phenotypes of coenzyme Q10-deficiency in the next-generation sequencing era