Role of transfer RNA modification and aminoacylation in the etiology of congenital intellectual disability
2020-07-30MartinFranzLisaHagenauLarsJensenAndreasKuss
Martin Franz, Lisa Hagenau, Lars R.Jensen, Andreas W.Kuss
Department of Functional Genomics, Interfaculty Institute for Genetics and Functional Genomics, University Medicine Greifswald, Greifswald 17475, Germany.
Abstract
Keywords: transfer RNA modification, aminoacylation, intellectual disability, aminoacyl-tRNA synthetases, ARS, human cognition, cognitive impairment, brain development
INTRODUCTION
Cognitive impairment features among the most important problems in healthcare, one prominent example being intellectual disability (ID) with a prevalence between 1% and 3%.The majority of severe forms of ID have specific yet very heterogeneous genetic causes, including numerous X-chromosomal as well as autosomal gene defects and disease-causing copy-number variants[1-5].Thus, with the exception of a few more prominent syndromes (for pertinent reviews see e.g., Salcedo-Arellanoet al.[6], 2020, Glassonet al.[7], 2020, Antonarakiset al.[8], 2020), individual genes only account for an often extremely low proportion of cases.
Accumulating evidence, however, indicates that while there are no major players on a genetic level, there are functional contexts or pathways that play a prominent role in the etiology of hereditary forms of ID and are thus of major importance for the development and maintenance of higher cognitive functions.One such feature is the molecular and functional integrity of transfer RNA (tRNA), and we and others have recently put forward the notion that a full as well as a fully functional complement of tRNAs is vital for human cognition[9,10].This is corroborated by the results of a survey of the recent literature, which shows a steep increase in the number of articles featuring tRNA-related issues in the context of impaired human cognition over the last few years [Figure 1].In support of the hypothesis that tRNAs play a major role in the basis of human cognitive features, our review aims to provide a synopsis of the presently available literature on tRNA modifiers and aminoacyl-tRNA synthetases (ARSs) that were found to play a role in the etiology of cognitive dysfunction.
tRNA STRUCTURE AND FUNCTION
tRNAs are important mediator molecules that facilitate the reading and translation process of the triplet genetic code from messenger RNA (mRNA) to corresponding polypeptides during protein biosynthesis[11].The human genome contains more than 500 tRNA genes[12]; however, tRNA expression is cell- and tissuespecific and approximately half of the genes are not or poorly expressed[13].
The typical tRNA secondary structure, consisting of hydrogen-bonded stems and associated loops, is shown in Figure 2.This results in a complex three-dimensional folding of the molecule, so that in their tertiary structure all tRNAs assume an L-shape.The 3’ end of this structure serves as the amino acid attachment site.The anticodon loop, which is exposed at the tip of the L-shape, is used for mRNA codon recognition.Base pairing with the first and third residue of the anticodon can be flexible so that some tRNAs can recognize various codons.
The translation of proteins from their coding mRNAs, where tRNAs play a central role, is an absolutely essential process.It begins with the formation of the pre-initiation complex, which is formed from the 40S subunit of a ribosome, the initiator tRNAMet, GTP and various initiation factors.mRNA binds to this complex at its 5’ end and translation is initiated when a start codon (AUG) is recognized.Elongation starts with the binding of the initiator tRNA to the peptidyl site of the ribosome, the second binding site of the ribosome, and the aminoacyl site is then occupied by the next tRNA.A peptide bond is formed between the methionine of the initiator tRNA and the amino acid of the following tRNA.The ribosome then moves one position further on the mRNA and binds another aminoacylated tRNA.This elongation continues until a stop codon is reached, after which the polypeptide leaves the ribosome[14].This happens at a rate of approximately ten tRNAs per second.
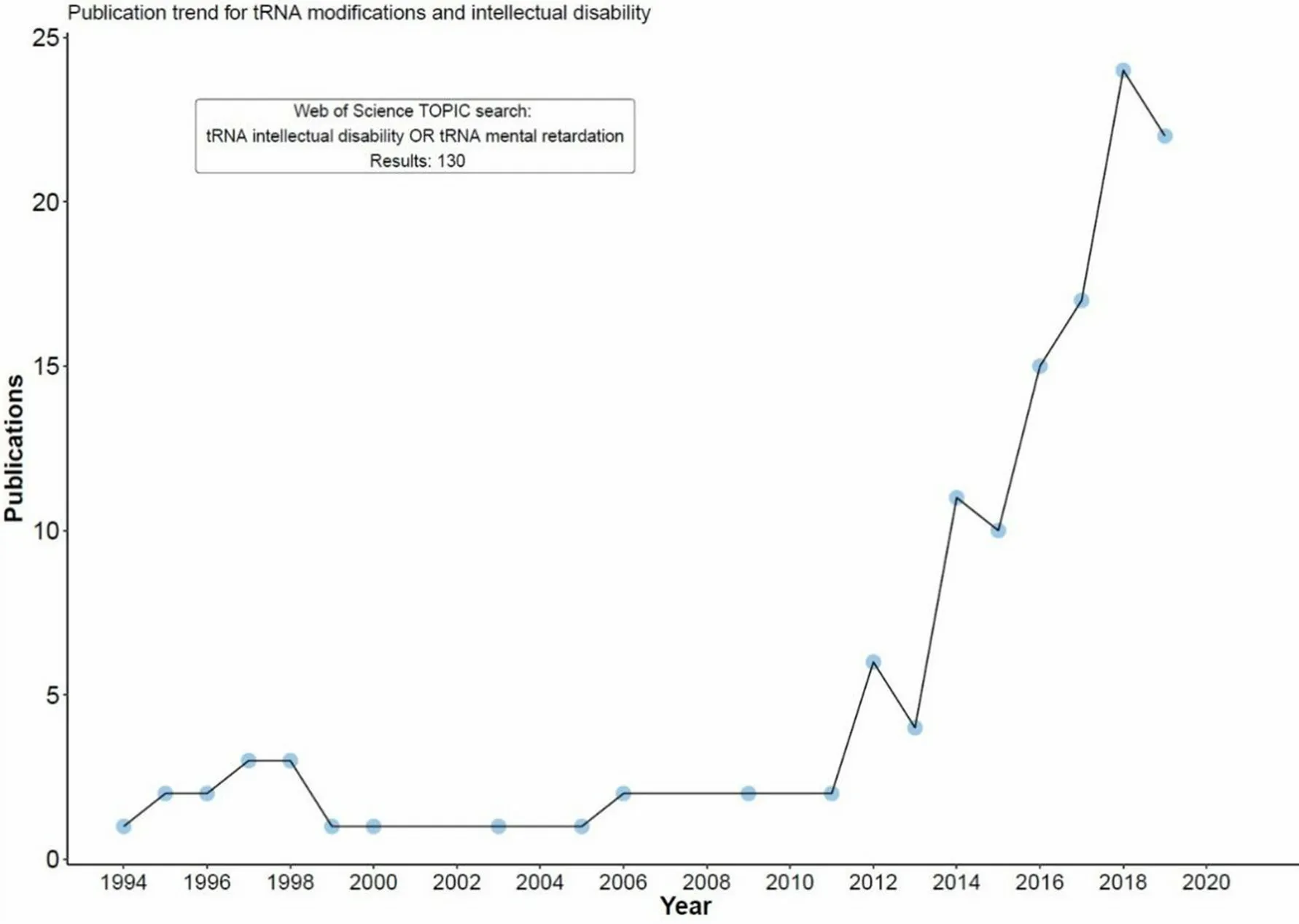
Figure 1.Number of articles featuring tRNA-related issues in the context of impaired human cognition between 1994 and the present

Figure 2.Overview of the main target nucleotides of the indicated tRNA modifiers involved in the etiology of ID.A-arm: acceptor stem; D-arm: dihydrouracil arm; C-arm: anticodon arm; ACL: anticodon loop; V-arm: variable arm; T-arm: ribothymidine arm; ID: intellectual disability
To ensure that protein synthesis runs smoothly, tRNA molecules are chemically modified[15-18].These alterations include methylation (guanosine → 7-methylguanosine), deamination (adenine → inosine), Sulfur substitution (uridine → 4-thiouridine), intramolecular rearrangements (uridine → pseudouridine) and the saturation of existing double bonds (uridine → dihydrouridine).Some of the non-standard ribonucleosides are believed to be important for tRNA stability and folding, or to improve codon-anticodon recognition[19-21].The wobble-uridine modification 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U), for example, was found to be associated with improving codon-anticodon recognition[22-25], and mcm5s2U plays a role in improving tRNA binding to the ribosomal aminoacyl-tRNA binding site[26].Without the modification, reduced binding at the aminoacyl-site leads to downstream effects, including slowing of the ribosomes and associated protein folding defects[23,27-29].Defects in tRNA modifications, which sometimes only represent a single atom, can trigger serious neurodegenerative diseases[30].For example, if tRNA molecules lack only a single chemical group, protein biosynthesis can stop at innumerable sites in the mRNA (reviewed in Torreset al.[31]2014).The result is an increase in protein aggregates that the cells can no longer remove.Nerve cells in particular are very sensitive to such aggregates, as is well known from Alzheimer’s and Parkinson’s diseases[27].Moreover, ribosome profiling experiments have shown that ribosomes in cells with defects in tRNAs take longer to read certain sections of the mRNA[27].The fact that protein biosynthesis does not occur at a constant rate plays a major role in this context, because changes in protein synthesis rate can influence protein conformation, as proteins take on their active form at the same time as they are produced[32].
Another important function in protein biosynthesis is performed by the ARSs.These enzymes are essential for translation, since they catalyze the binding of the proteinogenic amino acids to their respective associated tRNAs to form aminoacylated tRNAs.
There are 37 ARSs known - 17 occur only in the cytoplasm, 17 are mitochondria-specific, and three encode bifunctional proteins that charge tRNAs in both compartments[33].It is known that mutations in genes coding for ARSs play an important role in many human inherited diseases, both with recessive and dominant inheritance patterns.In homozygous carriers, recessive mutations in ARSs often cause early-onset disorders with a severe course, not only affecting nerve cells but also impairing the function of many other tissues.A total of 31 of the 37 human ARSs have been linked to a genetic phenotype.These range from later-onset peripheral neuropathy to severe multi-system development syndromes[34-36]with ID.
In the following sections, we will first give an overview of tRNA modifiers that have been found to play a role in the etiology of hereditary forms of cognitive impairment, focusing on the major tRNA sites targeted by these enzymes.Subsequently, we will introduce the ID-associated ARSs known to date, based on their cytosolic or mitochondrial occurrence.
TRNA MODIFICATION AND ID
A list of currently known tRNA modifiers, which have been associated with ID, is given in Table 1 (see Part A).The tRNA schematic in Figure 2 gives an overview over the main target nucleotides of tRNA modifiers involved in the etiology of ID, showing that there are 4 main sites that are of particular importance for human cognition: the C-arm (anticodon arm), V-arm (variable arm), D-arm (dihydrouridine-arm) and T-arm (ribothymidine arm).
Anticodon arm
The anticodon arm of a tRNA molecule contains the anticodon site and is the most heavily modified part of the tRNA molecule.
Currently, six different ID proteins catalyzing tRNA modifications in this tRNA region have been identified.These include the following enzymes.
FTSJ1
FTSJ1 (filamentous temperature-sensitive J,E.colihomolog 1) is an X-linked tRNA 2’-O-methyltransferase that catalyzes ribose methylation at tRNA positions 32 and 34.The homologous gene was originally isolated from anE.coliin 1991[178], and the crystal structure with the methyl donorS-adenosyl-methionine was later solved[179].
Yeast has been the model of choice for investigations concerning FTSJ1 as human FTSJ1 is able to complement yeast Trm7 growth defects[180].In yeast, two different interaction partners have been identified that are necessary for methylation at positions 32 and 34 in the anticodon loop, respectively.Trm7 interacts with Trm732 to methylate tRNAs encoding Trp (CCA), Phe(GAA) and Leu (UAA) at position 32 and with Trm734 to methylate at position 34 of tRNATrp(CCA), tRNAPhe(GAA) and tRNALys(cmnm5UmUU)[181].The tRNALysresidue cmnm5Um is a 5-carboxymethylaminomethyl 2’-O-methyluridine.The human homologs of Trm732 and 734 are THADA and WDR6, respectively.So far, WDR6 has not been found mutated in a M endelian disorder, whereas translocations disrupting THADA have been found in cer tain thyroid adenoma cells[182].However, the tRNA modification status in these cells has not been investigated.
Yeast is a good model for molecular investigations into tRNA modifications, but far from identical to the human situation.For example, yeast Trm7 methylates tRNAs encoding phenylalanine, leucine and tryptophan whereas human FTSJ1 methylates tRNAs encoding phenylalanine, asparagine, glutamine, alanine and methionine[181].
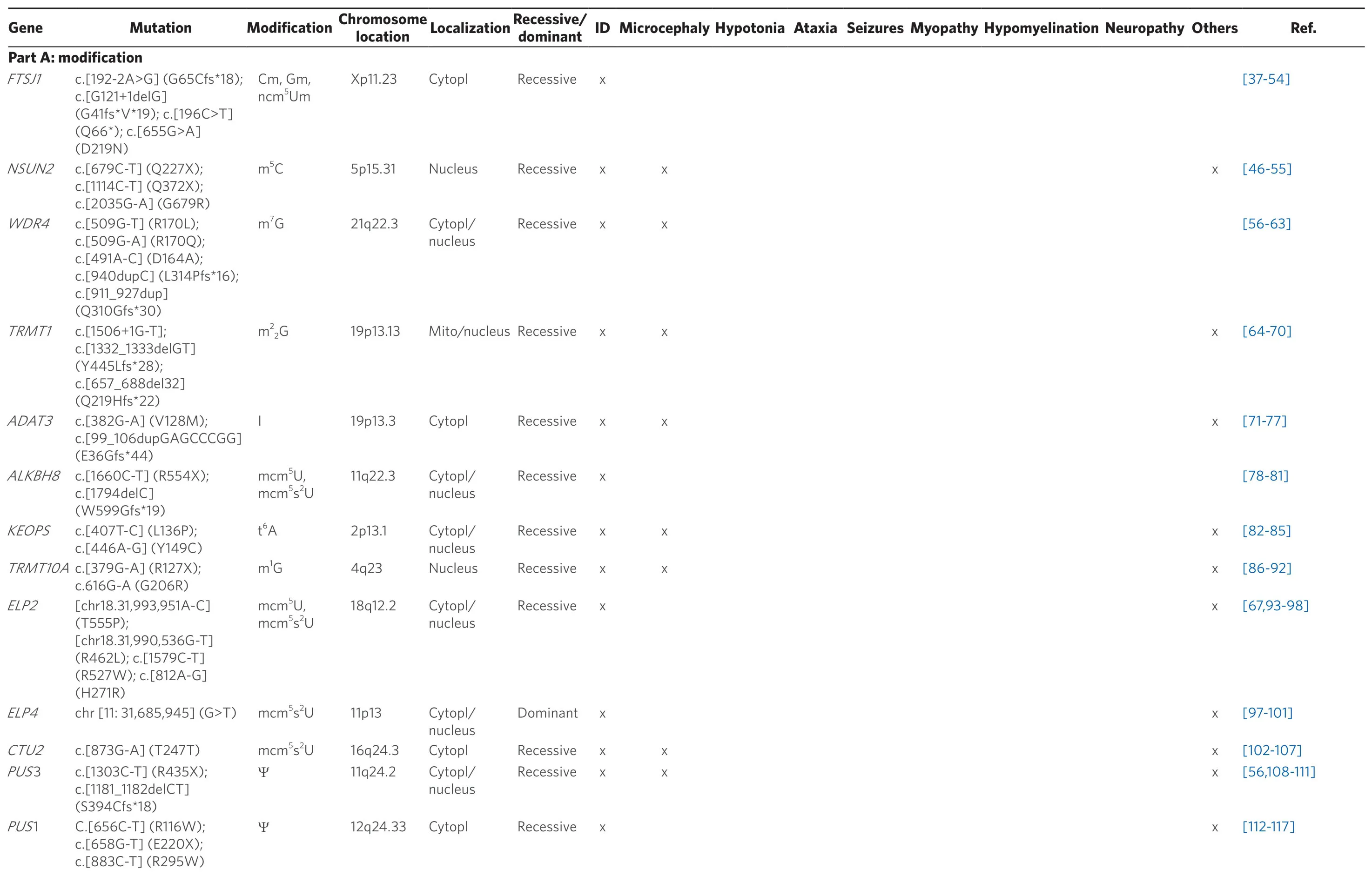
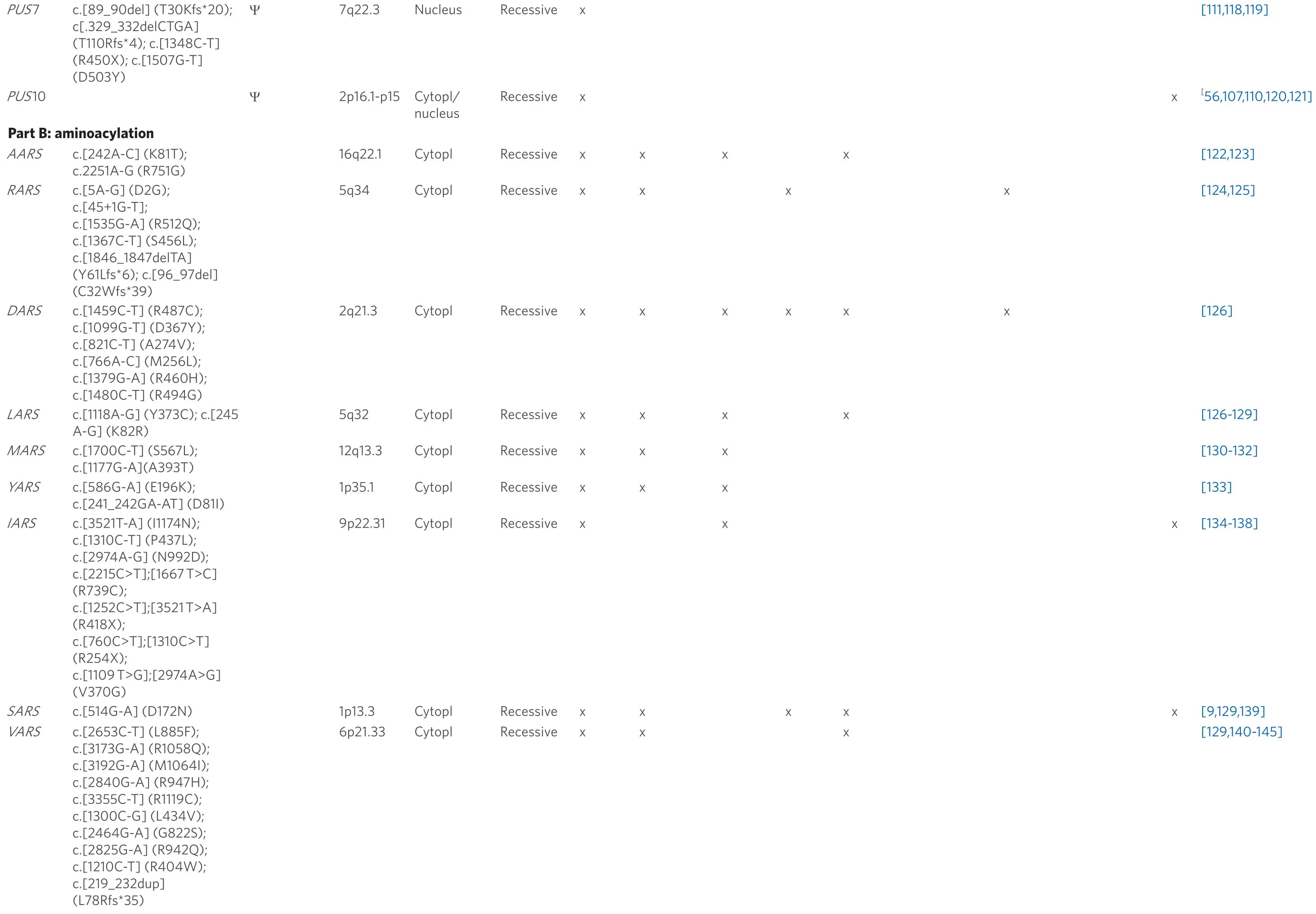
Table 1.List of currently known tRNA modifiers (Part A: modification) and aminoacyl-tRNA synthetases (Part B: aminoacylation) that have been associated with ID
?

cytopl: localization in cytoplasm; mito: localization in mitochondria; others: prenatal growth retardation, speech and hearing impairment, aggressive behavior, encephalocardiomyopathy, pontocerebellar hypoplasia, multiple respiratory chain complex defects, infantile onset developmental delay; abbreviations for tRNA modifications are according to the MODOMICS database[177]
In ID patients, protein-truncating mutations have been reported in 5 families[37,41,183], and a missense change has also been found in patients with non-syndromic ID[180].In addition, duplication or microdeletions involvingFTSJ1and other ID genes were also found in ID families[184-187].Recently, a mouse model forFTSJ1deficiency was reported.In combination with a mild ID phenotype, these mice presented with additional phenotypic features, some of which were also found in affected humans upon reexamination of patients who were previously considered to have a non-syndromic phenotype[40].
ADAT3
ADAT3 (adenosine deaminase TRNA specific 3) is part of an enzyme complex involved in inosine formation through hydrolytic deamination of adenosine at the tRNA wobble position.This protein was also first characterized in yeast and is specific for modification of the tRNA wobble position.In yeast, Adat3 complexes with Adat2 to function as a deaminase, and the coding genes for both are essential for yeast viability[73].In human cells, ADAT2 and 3 form a complex, localized in the nucleus that is required for inosine formation at the tRNA precursor level[188].
ID caused byADAT3mutations is inherited in an autosomal recessive manner and several consanguineous families have been analyzed mainly in the Middle East.ADAT3mutations were first identified in 24 individuals from eight consanguineous Arab ID families that all presented with ID and strabismus[71].The missense change c.382G>A, V128M, is located in an ancient haplotype that is approximately 1600 years old and considered to be the most common cause for autosomal recessive ID in Arabia[71].Other clinical symptoms in ID patients with theV128Mmutation apart from ID and strabismus were reported to include growth failure, microcephaly and tone abnormality[72].The authors concluded that despite a distinct facial profile, this syndrome should be considered also for ID patients from apparently non-consanguineous ID families originating from Arabia[72].Recently, an 8-bp duplication inADAT3was found in a patient with mild ID, microcephaly and hyperactivity but without strabismus[75].
In a patient cell line, it was recently shown that theADAT3mutationV128Mindeed reduces adenosine deaminase activity and inosine formation at the tRNA wobble position[74].
ALKBH8
ALKBH8 (alkylated DNA repair protein AlkB homolog 8) was originally investigated because it is expressed in human cancers, including bladder cancer[80].Knockdown of ALKBH8 in cell lines has shown several effects including reduced H2O2generation, induction of JNK- and p38-mediated apoptosis, phosphorylation of the histone 2 variant H2AX and reduced gall bladder cancer growth[80].In a mouse model for ALKBH8 deficiency, Alkbh8 was identified as a methyltransferase necessary for 5-methoxycarbonylmethyluridine (mcm5u) formation of wobble uridine residues[189].Generation of mcm5u is required for ALKBH8 hydroxylation of wobble uridine to 5-methoxycarbonylhydroxymethyluridine in certain tRNAs[78,190].Although Alkbh8-deficient mice seemed normal, the authors observed aberrant modification of selenocysteine-specific tRNASec[189].
Recently, truncatingALKBH8mutations were found in ID patients from two consanguineous families with different mutations (c.1660C>T, p.Arg554Ter and c.1794delC, Trp599GlyfsTer19).tRNA from the investigated patients showed complete loss of wobble uridine modifications.All seven investigated patients had ID and showed global developmental delay.Out of the seven patients, only one affected sister did not present with epilepsy[79].
CTU2
CTU2(cytosolic thiouridylase 2) is also a highly conserved gene that was first identified in yeast and found necessary for tRNA thiolation in yeast,C.elegansand even plants[102,104,191].In these organisms, CTU1 and CTU2 homologs form a complex that catalyzes tRNA thiolation of wobble uridine.Inactivation of the complex leads to loss of thiolation at the tRNA wobble uridine and abnormal phenotypes[102,104,191].Interestingly, proteins involved in thiolation of the uridine wobble base are also important for the altered protein synthesis driven by theBRAFV600Eoncogene transformation in melanomas, and melanomas depend on these tRNA-modifying proteins for survival[103].
The first humanCTU2mutations were reported in three families from Saudi Arabia and two families from the United Arab Emirates, and they were all homozygous for the same haplotype and splice site mutation (c.873G>A, Thr247AlafsTer21).The affected individuals presented with dysmorphic faces, renal agenesis, ambiguous genitalia, polydactyly and lissencephaly, and the authors suggested the acronym DREAM-PL for this syndromic form of ID[105,107].
Five more patients with the DREAM-PL phenotype were recently reported, all showing a reduced ratio of thiolated wobble uridine to unmodified wobble uridine[106].
KEOPS
KEOPS (kinase, endopeptidase and other proteins of small size) and the KEOPS complex were originally identified in yeast as a complex involved in telomere capping and elongation[192].In 2010, yeast KEOPS was found to be necessary for N6-threonyl-carbamoyl-adenosine modification of yeast tRNA adenosine (t6A), which is present at position 37 in all tRNAs that pair with ANN codons[193].Although telomere regulation seems to be independent of t6A modifications[66], yeast cells lacking t6A modifications show severe growth defects.
The human and yeast KEOPS complex each consist of four homologous subunits (OSGEP, TP53RK, TPRKB, LAGE3 and kae1, Bud32, Cgi121, Pcc1, respectively), and mutations were found in genes encoding any of the four subunits in Galloway-Mowat syndrome (GAMOS, MIM#251300) patients[83].These patients were all affected by early-onset nephrotic syndrome, primary microcephaly, developmental delay and propensity for seizures of which most patients died in early childhood[83].None of the patients carried truncating mutations on both alleles[83].
Due to the multiple functions involving the KEOPS complex it is difficult to determine the effect of a missing t6A modification on the patient phenotype.However, as overlapping phenotypes are observed in patients withWDR4mutations, missing t6A modifications are likely to contribute to the observed GAMOS phenotype[82].
ELP2 and ELP4
The Elongator protein complex (ELP) is composed of six highly conserved subunits (ELP1-6) and, as the name suggests, was initially thought to promote elongation of transcription.Recently, it was discovered that its primary role is to modify the uridine at position 34 of tRNAs (mcm5s2U)[194,195].Mutations in two of the Elongator subunits have been linked to ID.Missense mutations in theELP2gene have been identified in three families with ID[67,93].Microdeletions in theELP4gene have been linked to ID and speech delay, although deletion of part of the regulatory regions ofPAX6may contribute to the phenotype[99,100].Previously, mutations in theELP4gene have been implicated in Rolandic epilepsy[101].It is now accepted that the diverse disease phenotypes caused by defects in Elongator are likely due to hypomodified tRNAs, but it remains to be seen whether rescue experiments with elevated tRNA levels prevent the phenotypes in multicellular organisms[97,98].
PUS1, PUS3 and PUS7
Pseudouridine is a common tRNA modification, and to date, three ID proteins that play a role in pseudouridinylation have been identified.These are PUS1, PUS3 and PUS7 (pseudouridine synthases), which are involved in the conversion of uridine to pseudouridine at different specific tRNA positions.
Yeast Pus1 was the first eukaryotic tRNA pseudouridine synthase to be characterized and shown to be involved in the conversion of tRNA uridines at multiple positions of introns containing tRNAIle[196].Pus1 targets both cytoplasmic and mitochondrial tRNA[112]and was later shown also to target U2 snRNA in yeast[115].
The first reported humanPUS1mutation was a homozygous missense change (R116W) found in all affected individuals in two Italian families who suffered from mitochondrial myopathy and sideroblastic anemia (MLASA; MIM 600462) but without ID[113].tRNA pseudouridinylation was later shown to be greatly reduced in patient cell lines[116].PUS1-dependent ID was first reported in a patient with the same (R116W) missense change by Zehariaet al.[117].In two brothers with MLASA and a truncatingPUS1mutation (E220X), one had ID whereas the other had an elevated intelligence quotient above normal levels[114].
PUS3 is a pseudouridine synthase, originally isolated from yeast, that catalyzes pseudouridine formation at positions 38 and 39 in the anticodon stem of certain tRNAs.Yeast Pus3 deletion strains are viable but grow slowly, especially at elevated temperatures[197].The protein was found to be evolutionarily conserved, and like mouse Pus3, it can convert uridine at position 38 or 39 to pseudouridine in yeast and human tRNAin vitro, albeit with different efficiency[109].
ID caused by PUS3 deficiency is inherited as an autosomal recessive disorder.The first report ofPUS3mutations described 3 affected sisters that were homozygous for the nonsense mutation c.1303C>T, R435X, and the phenotype in these patients was largely brain specific[110].A second report presented a single child from consanguineous parents, carrying a frameshift mutation (c.1181_1182delCT, Ser394CysfsTer18) and no detectable PUS3 transcript.The child suffered from ID, microcephaly, hypotonia, seizures, and vision and hearing loss[56].Furthermore, two compound heterozygous mutations were reported in a Brazilian and a Chinese family[198,199].Although all reported patients presented with additional features, ID was the only consistent characteristic.
PUS7 is a multi-substrate pseudouridine synthase that in yeast targets several tRNA uridines at position 13, the pre-tRNATyrat position 35[200], small nucleolar RNA U2 (U2 snRNA) at position 35[201]and also 5S and 5.8S rRNA[202]and mRNA[203].Interestingly, uridine conversion of snRNA U2 at positions 56 and 93 can be induced in yeast by nutrient deprivation or heat shock[204].In human stem cells, PUS7 pseudouridinylation was found to activate small tRNA-derived fragments that inhibit protein synthesis by targeting the initiation complex.PUS7 inactivation leads to defective germ layer specification[205].
Homozygous truncatingPUS7mutations were recently reported to cause ID with speech delay, short stature, microcephaly, and aggressive behavior in patients from three different families[118].Two ID families with homozygousPUS7mutations, a missense change or a deletion leading to a frameshift, were also reported.The patients also suffered from microcephaly, whereas short stature was not seen in all patients[111].Recently, another ID family of Afghan origin was reported, carrying a Gly128Arg missense change.The phenotype of the patient was milder without microcephaly or short stature, but still with speech delay and aggressive behavior[119].In this last study, pseudouridine levels were not investigated[119], whereas markedly reduced pseudouridine levels at tRNA position 13 were found in all investigated ID patients[111,118].
Variable arm
The variable arm of tRNAs is located between the anticodon (or C) and the T arms.The length of the variable arm depends on the tRNA and can be between 3 and 21 nucleotides long.Generally speaking, class I tRNAs have shorter variable arms (between 4-5 nucleotides) than class II tRNAs (> 10 nucleotides)[206,207].The variable arm functions as a stabilizer of the tertiary structure as well as in the specific recognition of the ARS.So far, two modifications of nucleotides in the variable arm by two different genes have been linked to ID.
NSUN2
NSUN2 (Nop2/Sun RNA methyltransferase family member 2) is one of three cytosine-5 tRNA methyltransferases and is responsible for methylating tRNAs that carry a cytosine at positon 48 or 49.There have been several reports linking mutations in theNSUN2gene to ID[46,49,51,208].Two reports observed a Dubowitz-like syndrome in patients[46,51].Other common symptoms described include microcephaly, facial dysmorphism and growth retardation.
The likely molecular mechanism in NSUN2-deficient cells is increased angiogenin-induced fragmentation of tRNA which inhibits protein translation[55].Methylation of cytosine at the variable loop in healthy cells protects tRNAs from binding to angiogenin.
WDR4
WDR4 (WD repeat domain 4) encodes the noncatalytic subunit of the tRNA (guanine-N7-)-methyltransferase which is necessary for the 7-methylguanosine modification (m7G) at position 46[62].It has been described to cause primordial dwarfism, a phenotypically diverse syndrome with several subtypes, characterized by ID as well as pre- and postnatal growth deficiency[58,62,63].More recently, WDR4 deficiency has also been linked to the Galloway-Mowat syndrome[57].WDR4 knockouts result in a complete loss of m7G modification in tRNAs and consequently to disturbed codon recognition and ribosome stalling.It has also been shown that depletion of WDR4 in mice impairs the neural lineage differentiation capacity in mESCs[60].
D-arm
The D-arm of tRNAs is located between the anticodon and acceptor arms.It is of variable length, but the modification of the D-loop nucleotides is highly conserved in all kingdoms.Its function is mainly the stabilization of tRNA structure through tertiary interaction with the T-arm, but it is also involved in aminoacyl tRNA synthase recognition.Defects in two tRNA methyltransferases that modify different positions in the D-arm have been shown to cause ID.
TRMT1
TheTRMT1(TRNA methyltransferase 1) gene encodes for a tRNA methyltransferase that dimethylates G at position 26 in the D-arm of most tRNAs.It was first connected to non-syndromic ID in a deep sequencingbased screen for novel genes for cognitive disorders in 2011[67].More recent reports confirm this finding and describe facial dysmorphism, general developmental delay and in some cases muscle weakness and spasticity as TRMT1-specific symptoms in patients[64,65,70].Apart from decreased protein translation and cell proliferation, TRMT1-deficient cells show disturbed redox homeostasis and hypersensitivity to oxidative reagents, which might explain some of the neurological defects observed[69].The causative mechanism at the tRNA level is still unclear; however, loss of m22G could affect tRNA structure or stability[209,210]or modulate translation activity[211].
TRMT10A
TRMT10A (TRNA methyltransferase 10A) is a tRNA methyltransferase that is responsible for methylating the G at position 9 of tRNAs (m1G9).A missing, shortened or otherwise non-functioningTRMT10Agene causes ID, microcephaly and general developmental delay[86].Interestingly, some reports describe early-onset diabetes or hypoglycemia in patients with mutations in the gene[86,88,91,92].
A lack of m1G9modification in yeast has been shown to play a role in tRNA stability and translation terminating efficiency[212,213].In human tRNALys, which has an adenine at position 9, a lack of methylation prevented the tRNA to be folded into the cloverleaf form[214].However, how exactly the lack of methylation is connected to the variety of symptoms is still not fully understood and continues to be the subject of ongoing research.
T-arm
PUS10
So far, no modifications on the T-arm of tRNAs have been shown to cause ID specifically.While microduplications or -deletions of the 2p16.1p15 locus, which contains the pseudouridine synthase 10 gene (PUS10) among several other genes, have been linked to ID and developmental and speech delay[215-217], there is growing evidence that in these cases, BCL11A is the cause for ID[218,219].Still, it cannot be ruled out that PUS10, which pseudouridinylates tRNAs at positions 54 and 55, contributes to the phenotype, but clinical cases with PUS10-specific mutations linked to ID have not yet been described so far.
ARSS AND ID
Cytoplasmic ARSs
The main task of ARSs is to transfer and bind amino acids to the appropriate tRNA molecules.The charged tRNAs are then used by the ribosomes to carry out protein synthesis.Their availability therefore plays an essential role in the regulatory processes of cell functions[220].All ARSs are ubiquitously expressed and highly conserved.There is one ARS enzyme for each amino acid to facilitate binding with the appropriate tRNA.Of the 37 knownARSgenes, 17 encode purely cytoplasmic enzymes[33].Like mitochondrial ARSs (mt-ARSs), all cytosolic ARSs (ct-ARSs) are encoded by nuclear genes.They are complemented by three ARSs that function in both the cytoplasm and mitochondria to match the full complement of amino acids.It has already been mentioned that biallelic mutations in 31 ARS genes lead to serious recessive, early onset diseases, ranging from later-onset peripheral neuropathy to severe multi-system development syndromes.Here, however, we will focus only on ARSs, which have been found to play a role in the etiology of diseases associated with ID [Table 1, see Part B].
InVARS,for example, Friedmanet al.[140]found different biallelic mutations in several families, leading to a very heterogeneous symptomatic picture including, developmental delay, epileptic encephalopathy and primary or progressive microcephaly.Another interesting case is the glutaminyl-tRNA synthetase gene (QARS).This gene encodes both the cytosolic as well as the mitochondrialQARSand shows a strong level of expression in the brain of the developing fetus.A very often found missense mutation (V476I) inQARSwas shown to cause a reduction in its aminoacylation activity[148].Mutations inQARShave severe consequences in affected individuals including not only ID but also progressive microcephaly, cerebral cerebellar atrophy and seizures that are difficult to treat.Altogether 11 patients have so far been described withQARSmutations[147,149,151,221], all of whom consistently show a severe so-called global development delay but none reaching any significant milestone.An initially normal occipito-frontal circumference (OFC) quickly and clearly changed to postnatal microcephaly.Various degrees of severity of ID from mild to severe were described in several case studies [Table 1B].In addition to other serious symptoms, the condition is ultimately fatal for a large proportion of patients[148].These examples shows the breadth and variability of the phenotypic spectrum associated withARSmutations.
There are, however, recurrent motives among the features accompanying ARS-dependent ID, such as microcephaly, which is observed in carriers of mutations inAARS, RARS,DARS, LARS, MARS, YARS, QARS, SARS, VARSandWARS2[Table 1B].An association with the occurrence of seizures (AARS, DARS, LARS, SARS, VARS, QARS, NARS2, PARS2 and WARS2) and hypotonia (AARS,DARS, LARS, MARS, YARS and IARS) is also frequently observed.Less common features among affected individuals range from ataxia, cerebral atrophy, neonatal choleastasis, muscular hypotension, infantile hepatopathy and hypomyelination to speech disorders and aggressive behavior.Finally, it should be mentioned that the non-canonical functions of ARSs could also be responsible for the wide phenotypic spectra that can be observed in the diseases related to their mal- or dysfunction.
Mitochondrial ARSs
Human mitochondrial ARSs (mt-ARSs) are essential for the synthesis of 17 mt-DNA-encoded proteins, which are all subunits of the respiratory chain complexes.Therefore, they are involved in the generation of the major source of cellular energy, i.e., ATP.Like cytosolic ARSs, all mt-ARSs are encoded by nuclear genes, which are, however, different from those coding for the cytosolic ARSs.ThreeARSgenes encode enzymes that are active in both mitochondria and cytosol: glycyl-tRNA synthetase (GARS), lysyl-tRNA synthetase (KARS), and glutaminyl-tRNA synthetase (QARS).Only QARS, however, has so far been found to be associated with an ID phenotype [Table 1B].The first correlation between an mt-ARS mutation and a human disorder was published in 2007 by Scheperet al.[222], who found autosomal recessive mutations in theDARS2gene in individuals suffering from leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation (LBSL).Since then, numerous other pathogenic mutations in mt-ARSs have been described, so that to date, at least 17 out of the 19mt-ARSsgenes have been implicated in human genetic disorders involving damage to the central nervous system[35].
It is noteworthy at this point that in 2017, Moulinieret al.[223]introduced MiSynPat, an integrated knowledge base that links clinical, genetic, and structural data for disease-causing mutations in humanmt-ARSs.According to the authors, this tool provides a “comprehensive knowledge base together with an ergonomic Web server designed to organize and access all pertinent information (sequences, multiple sequence alignments, structures, disease descriptions, mutation characteristics, original literature) (http://misynpat.org/misynpat/AboutMisynpat.rvt last accessed 2020-01-09).
Mutations in at least sixmt-ARSgenes (Table 1B - aminoacylation, includingQARS) are involved in the etiology of ID.All of these lead to a syndromic phenotype.Mutations inNARS2andPARS2,for example, cause Alpers syndrome, and homozygousRARS2defects lead to pontocerebellar hypoplasia, which is characterized by not only overall delayed development, impaired brain development, movement problems and ID but also progressive atrophy, particularly of the pons and cerebellum.WARS2mutation carriers show a phenotype that is very similar to patients with mutations in cytosolicSARS(Table 1B - aminoacylation).Other than that seen for ct-ARSs, there are no clearly prominent recurrent motives in homozygous or compound heterozygous carriers of mt-ARS mutations (Table 1B - aminoacylation) with the possible exception of seizures that are observed with a notably increased frequency (NARS2,PARS2andQARS).
CONCLUSION
The literature compilation we present here makes a compelling case for an important if not pivotal role of a fully functional tRNA complement for the development and maintenance of higher cognitive functions.Interestingly, disease-causingARSsmutations often only result in a reduction of enzyme activity without causing complete inhibition[158,224,225].This points to the sensitivity of cognitive features towards even slight disturbances in this basic cellular process.
In addition, there is much evidence that tRNA molecules assume possibly unknown biological functions in eukaryotes, which have not yet been fully elucidated[17]but could be influenced by disruption of tRNA function.This opens up a myriad of further possibilities for tRNA involvement in the formation of cognitive features and underlines the importance of further research in this field.
DECLARATIONS
Authors’ contributions
Made substantial contributions to the conception and design of the article, performed literature research and interpretation and were involved in the writing and editing of the manuscript as well: Franz M, Hagenau L, Jensen LR, Kuss AW
Franz M and Hagenau Lcontributed equally to the article.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2020.
杂志排行

Journal of Translational Genetics and Genomics的其它文章
- Intellectual disability, the long way from genes to biological mechanisms
- Spectrum of MECP2 mutations in Indian females with Rett Syndrome - a large cohort study
- The North American mitochondrial disease registry
- Mitochondrial translation defects and human disease
- Cryogenic electron paramagnetic resonance spectroscopy of flash-frozen tissue for characterization of mitochondrial disease
- Redefining infantile-onset multisystem phenotypes of coenzyme Q10-deficiency in the next-generation sequencing era