小麦纹枯病抗性QTL分析
2020-07-30周淼平姚金保杨学明余桂红
周淼平,姚金保,杨学明,张 鹏,余桂红
(江苏省农业科学院粮食作物研究所,江苏南京 210014)
小麦纹枯病(wheat sharp eyespot,WSE)是发生在温带地区的世界性病害,在欧洲、北美洲、非洲、大洋洲和亚洲均有发生[1]。在中国,该病害主要由禾谷丝核菌(RhizoctoniacerealisVan der Hoeven)侵染小麦茎秆基部引起[2],侵染后,造成茎秆基部腐烂,小麦抗倒伏能力下降,由于病原菌菌丝堵塞,植株上部营养和水分的输送发生困难,严重时发生死苗和枯白穗,导致产量损失。据报道,中国每年小麦纹枯病发生面积超过600万公顷,仅2005-2008年,由于该病害造成的损失超过10亿元[3]。培育和使用抗病品种是减轻小麦纹枯病危害最经济、环保和有效的方法。
遗传分析表明小麦对纹枯病的抗性是多基因控制的数量性状[4-5]。分子数量遗传学的发展,使得定位和聚合小麦纹枯病抗性QTL成为可能。迄今为止,已经对小麦纹枯病抗源ARz[6]、川35050和山农0431[7]、白免3号[8]、AQ[5]、Niavt14[9]和CI12633[10]的抗性QTL进行了初步定位,在1A、2B、2D、3B、3D、4A、4B、5A、5B、5D、6A、6B、7B和7D染色体上均发现抗性QTL,但大多QTL表型解释率较低,且未发现抗性稳定的主效QTL,因此,继续筛选抗病种质资源、挖掘新的抗性QTL以及微效抗性QTL的聚合将是今后小麦纹枯病研究的主要内容[5]。
已报道的小麦纹枯病抗性QTL定位研究大多采用SSR标记,但该标记的多态性低,构建的遗传连锁图对小麦基因组的覆盖度较差,影响抗性QTL的检出效率。SNP芯片技术和高通量测序技术的发展为高密度遗传连锁图的构建提供了便利,Wu等[10]采用90K SNP芯片对CI12633/扬麦9号重组自交系群体进行了基因型分析,构建了12 434.23 cM的高密度遗传连锁图,定位了5个有关小麦纹枯病抗性的QTL位点,但这些QTLs位点在不同试验中的表型解释率总和仍然偏低,只有22.2%~63.7%,表明仍有不少纹枯病抗性QTL位点未被检出。
本研究以CI12633/扬麦158重组自交系群体中的94个家系为材料,采用二代测序技术开发SNP新标记,构建高密度遗传连锁图,结合牙签接种和病麦粒接种的方法鉴定重组自交系群体的纹枯病抗性,进行小麦纹枯病抗性QTL分析,以期获得更多的抗性QTL位点,为今后解析小麦纹枯病抗性机制以及开展抗小麦纹枯病标记辅助育种提供帮助。
1 材料与方法
1.1 试验材料
以感纹枯病品种扬麦158(江苏省里下河地区农业科学研究所提供)为母本,抗纹枯病品种CI12633(江苏省农业科学院种质资源与生物技术研究所提供)为父本,配制杂交,采用单粒传的方法构建重组自交系群体,该群体含有198个家系,分析时已达F8代,本研究对其中94个家系进行了基因型分析和纹枯病抗性鉴定。
禾谷丝核菌R0301由江苏省农业科学院植物保护研究所陈怀谷研究员提供,该病菌为江苏麦区致病力较强的小麦纹枯病病原菌。
1.2 重组自交系群体遗传连锁图的构建
重组自交系群体的亲本和各家系的叶片DNA提取参照Saghai-Maroof等[11]的方法。
SSR标记分析:根据GrainGenes网站提供的SSR标记引物序列(https://wheat.pw.usda.gov/cgi-bin/GG3/browse.cgi?class=marker)合成引物,在亲本间筛选多态性SSR标记,用于重组自交系群体各家系的基因型分析。
SNP标记分析:采用ddRAD-seq(double digest restriction site-associated DNA sequencing)技术开发SNP标记,重组自交系家系和亲本的基因组DNA分别采用限制性内切酶EcoRⅠ和NlaⅢ酶切并连接EcoRⅠ接头和NlaⅢ接头,琼脂糖凝胶电泳后回收400~600 bp连接产物,DNA定量后,等量混合24个样品,采用Illumina TruSeq DNA Library Prep Kits进行DNA文库构建,测序。测序数据处理后,通过reads间聚类方法以及与小麦参照基因组的比对,搜寻在亲本间存在的多态性ddRAD标签,开发SNP标记,并在重组自交系各家系进行分析。
根据重组自交系群体的SSR和SNP 分析结果,采用JoinMap 4.0软件构建各染色体遗传连锁图。
1.3 重组自交系群体的纹枯病抗性鉴定
重组自交系群体的纹枯病抗性鉴定分别采用牙签接种法和病麦粒表土接种法进行。
牙签接种法:牙签置于烧杯等容器内,清水冲洗浸泡过夜,沥干,加入已融化的PDA固体培养基,浸没牙签1/2,灭菌并凝固后,接种新鲜培养的禾谷丝核菌R0301,25 ℃黑暗培养,菌丝长满牙签后备用。于小麦拔节期接种,取带菌牙签,插于叶鞘与茎秆之间,小麦弥雾保湿7 d,每个重组自交系家系和亲本接种50个茎秆,于小麦乳熟期调查纹枯病病级,计算平均病情指数,每个试验重复2次。牙签接种于2005-2007年在江苏南京江苏省农业科学院本部试验地连续3次试验。
病麦粒接种法:小麦籽粒清水浸泡24 h,沥干,装袋灭菌,采用1 cm见方长有禾谷丝核菌R0301的PDA琼脂培养基接种,25 ℃培养,2 d翻动1次小麦籽粒,直至籽粒均匀布满菌丝备用。于小麦返青期接种,在小麦行间划浅沟,均匀撒播病麦粒后覆土,每天早晚喷水1次,连续7 d,病麦粒用量150 kg·hm-2。于小麦乳熟期调查纹枯病病级,每个家系和亲本调查50个茎秆,计算平均病情指数,每个试验重复2次。病麦粒接种分别于2006年在江苏扬州里下河地区农科所试验地、2012-2014年在江苏南京江苏省农业科学院本部试验地进行。
小麦纹枯病病级按以下标准确定:0级,叶鞘和茎秆均未见纹枯病病斑;1级,叶鞘有典型纹枯病病斑,但不侵茎;2级,病菌侵入茎秆,但病斑环茎宽度不超过茎秆周长的1/2;3级,病斑环茎宽度是茎秆周长的1/2~3/4;4级,病斑环茎宽度绕茎秆3/4以上,或茎秆已软腐;5级,枯孕穗或枯白穗。
病情指数按以下公式计算:病情指数=[(Σ各级病株数×各级代表值)/(总株数×最高级代表值)]×100%
1.4 数据分析和QTL定位
重组自交系群体和亲本纹枯病抗性鉴定的数据平均值和频率分布、试验间相关性和方差分析均采用R软件计算。
QTL定位采用MapQTL 5.0软件进行,先进行QTL区间作图(interval mapping),然后采用软件筛选的辅因子(cofactors)进行多QTL作图(MQM Mapping),根据置换检测(permutation test)推荐的LOD值判定是否存在QTL。
2 结果与分析
2.1 遗传连锁图的构建
经亲本多态性筛选和群体基因型分析,共有430个多态性SSR标记参与遗传连锁图构建。采用ddRAD-seq技术共开发SNP多态性标记有 11 965个,剔除群体缺失数据超过5%以及共分离的标记,共有4 086个多态性SNP标记参与遗传连锁图的构建。JoinMap 4.0软件构建的遗传连锁图含有31个连锁群,均可分配到相应的染色体,总遗传距离2 510.66 cM,含有分子标记 3 355个(表1)。
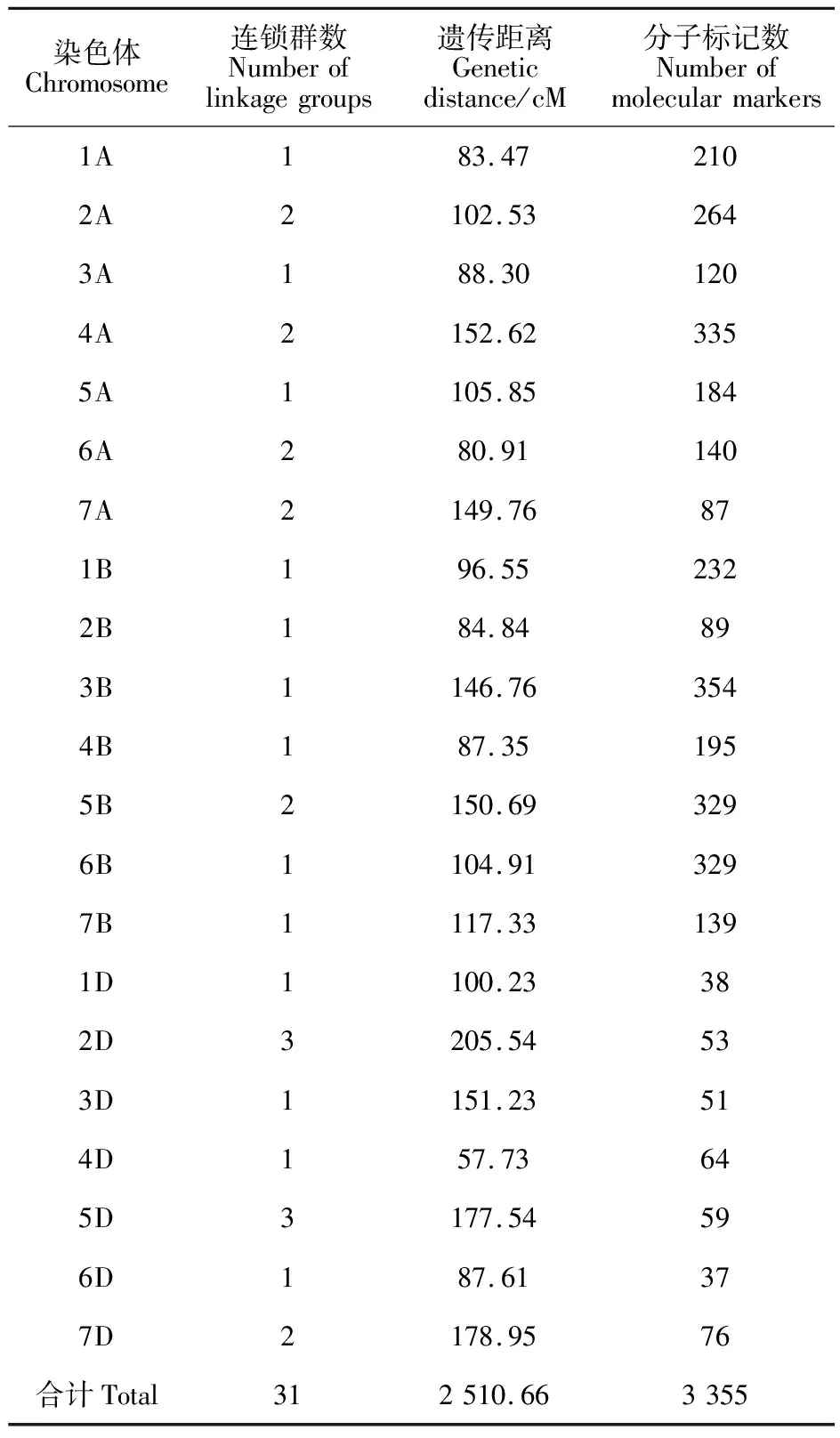
表1 群体遗传连锁图概况
2.2 重组自交系群体的纹枯病抗性鉴定
重组自交系群体和亲本的纹枯病抗性采用两种方法进行,牙签接种(YQ)分别于2005年(05YQ)、2006年(06YQ)和2007年(07YQ)在江苏南京试验地进行,病麦粒接种(BT)分别于2006年在江苏扬州(06YZ)、2012年南京(12NJ)、2013年南京(13NJ)和2014年南京(14NJ)进行。群体7次试验的偏度(skewness)和峰度(krutosis)值均接近于0,表明群体的病情指数基本呈正态分布,小麦纹枯病的抗性由多基因决定,群体中均有抗病和感病的超亲家系出现。三年牙签接种的群体平均病情指数为34.46%,平均变化范围在 23.54%~53.09%;四年病麦粒表土接种的群体平均病情指数为27.19%,平均变化范围在 18.10%~39.22%,病麦粒接种与牙签接种相比,群体的纹枯病发病程度偏轻(表2)。
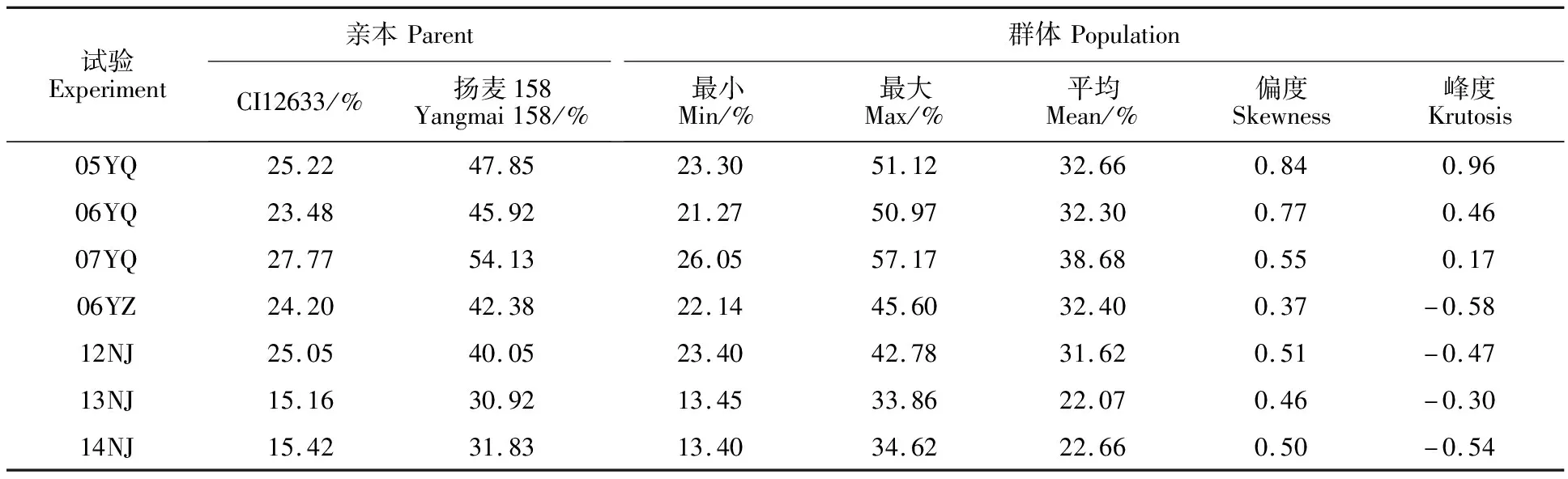
表2 群体纹枯病抗性鉴定概况
方差分析表明,牙签接种中,群体的各家系间以及试验间的方差达到极显著水平,但家系与试验的互作效应不显著;病麦粒接种中,各家系间、试验间以及家系与试验互作的方差均达到极显著水平(表3)。
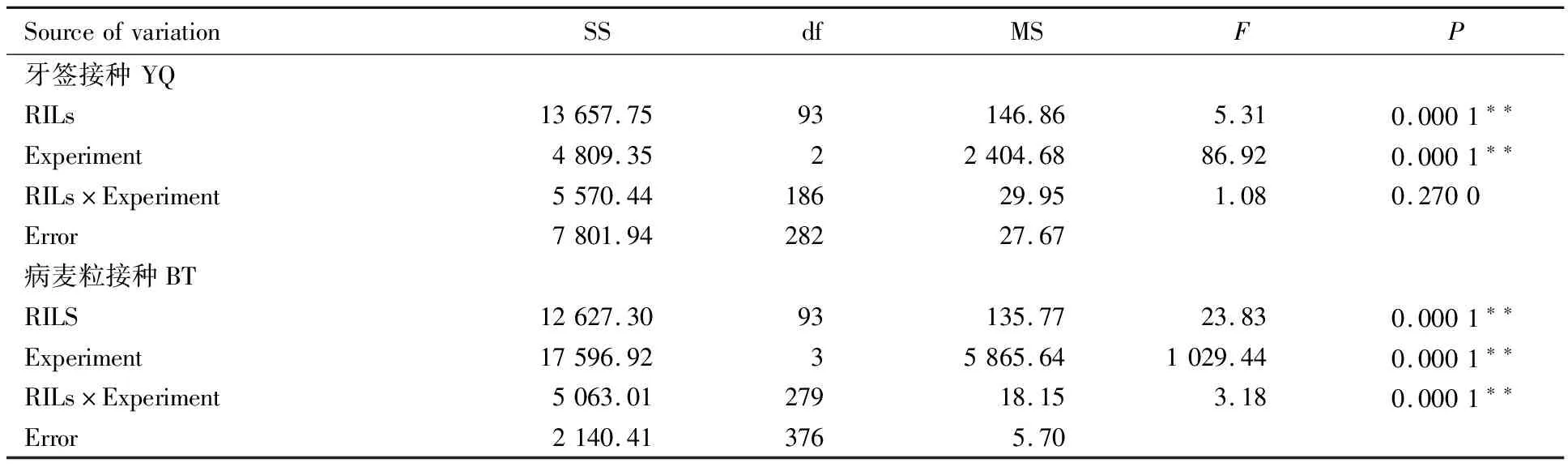
表3 试验的方差分析
2.3 纹枯病抗性鉴定试验间的相关性
7个试验间的相关系数在0.21~0.81之间,06YZ与牙签接种试验间的相关系数较低;牙签接种试验间平均相关系数为0.51,变化在0.34~0.71之间;病麦粒表土接种间平均相关系数为 0.66,变化在0.33~0.81(表4)。
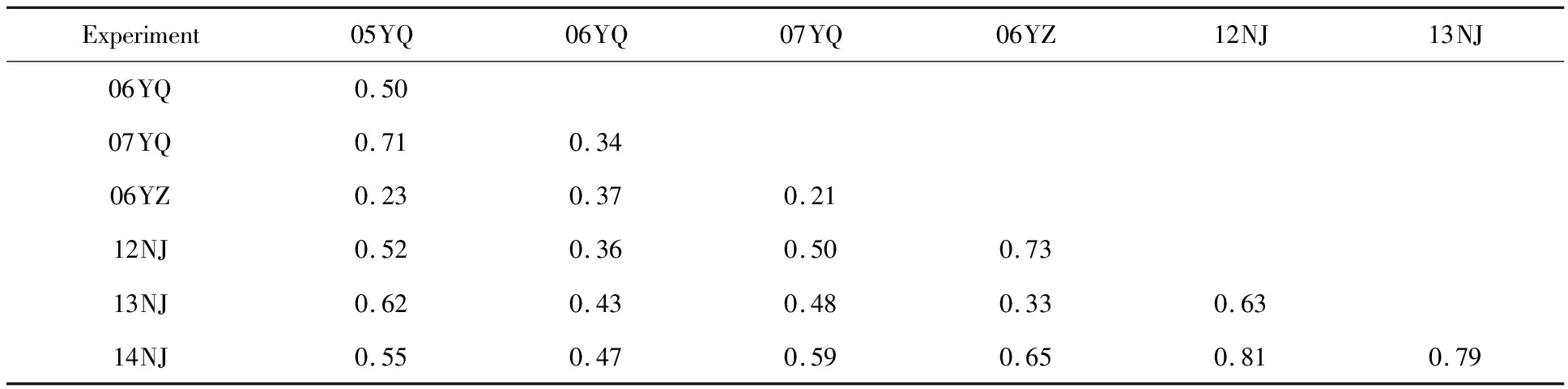
表4 试验间相关分析
2.4 纹枯病抗性QTL定位
2.4.1 牙签接种方法
采用牙签接种方法,共检测到8个纹枯病抗性QTL位点,涉及8条染色体,全部来自于抗病品种CI12633,但QTL位点的稳定性不高,未能检测到三年均表现一致的QTL,表明小麦纹枯病的侵染和扩展受环境因素影响较大。5A、1B和7D染色体上的抗性QTL在2年试验中均能检测到,其余3B、4B、5B、6B和4D染色体上的抗性QTL只在1年试验中检测到。定位的抗性QTL多为微效QTL,抗性表型解释率在10.80%~26.80%之间,其中7D的表型解释率最高,两年分别为26.80%和 22.90%,为纹枯病抗病主效QTL(表5)。
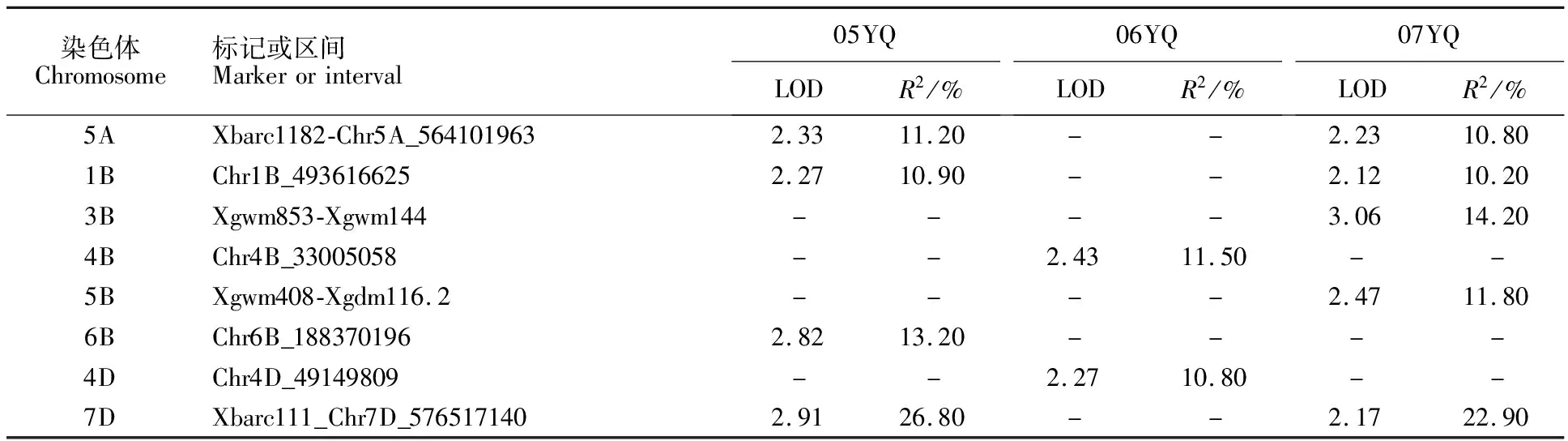
表5 牙签接种方法获得的抗纹枯病QTLs
2.4.2 病麦粒接种方法
采用病麦粒接种法,共检测到10个纹枯病抗性QTL位点,涉及9条染色体,其中2D染色体检测到2个抗性QTL位点;位于6B和1D染色体的QTL在4年试验中均能检测到,为纹枯病抗性稳定QTL;6A染色体上的QTL在3年试验中均能检测到;5A、2B、7B和2D染色体的QTL可在2年试验中检测到;其余位于1B、4B和2D染色体抗性QTL只能在1年试验中检测到。除了7B染色体上的QTL由感病品种扬麦158贡献外,其余QTL均来自抗病品种CI12633。所检测到的纹枯病抗性QTL表型解释率都较低,在9.10%~16.90%之间(表6)。
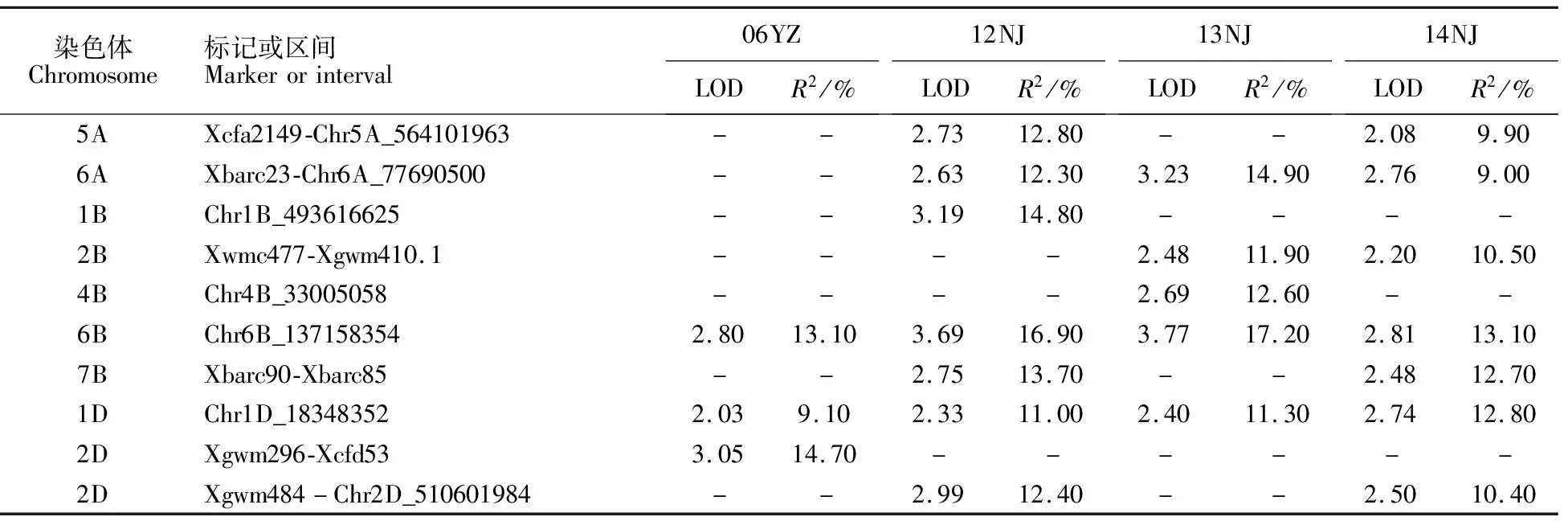
表6 病麦粒接种法获得的抗纹枯病QTLs
采用牙签接种方法和病麦粒接种方法均能在1B和4B染色体检测到的QTL位点,推测这两个QTL位点在小麦抗纹枯病中发挥重要作用。
3 讨 论
近几年随着高通量分子标记分析技术的进步,使小麦等作物重要性状的QTL定位发展迅速,群体基因型的分析时间缩短、遗传连锁图的分子标记密度加大,QTL定位的精确度愈来愈高。特别是小麦全基因组测序和物理图谱构建的完成,为小麦重要性状QTL的定位、比较和功能候选基因的确定提供了极大方便。本研究采用二代测序方法,参照发表的小麦基因组物理图谱,在CI12633/扬麦158群体中开发了11 965个SNP多态性分子标记,选用其中的4 086个开发标记并结合已有的SSR标记,构建了含3 355个标记、遗传距离为2 510.66 cM的小麦遗传连锁图,为小麦纹枯病抗性QTL的定位奠定了基础。
前人的研究中,小麦纹枯病抗性QTL定位多以SSR标记为主,单个研究由于基因组覆盖度不高,检测到的QTL数量较少。本研究采用SNP高通量分子标记,通过牙签接种和病麦粒接种两种方法,分别检测到8个和10个小麦纹枯病抗性QTL,涉及13条染色体,其中B组染色体均含有纹枯病抗性QTL,这也与以往B组染色体抗性QTL检出率较高结果相符[5-10]。将本研究检出的QTL与已报道的纹枯病抗性QTL在小麦基因组物理图谱上进行比较,发现5A、3B和7D染色体的QTL分别与Wu等[10]、Chen等[5]和汤颋等[6]报道的相应染色体上的QTL位置接近,可能为同一QTL,检出的其余QTL与已报道相应染色体的QTL位置不一致,可能为新检出QTL。
本研究检出的16个抗纹枯病QTL中,除了7B染色体的QTL由感病品种扬麦158贡献外,其余全部来自抗病亲本CI12633。Wu等[10]采用CI12633/扬麦9号重组自交系群体定位了4个来自CI12633的抗纹枯病QTL,分别位于4BS、5AL(2个)和5BS染色体,本研究只检测到5AL.1 QTL,其余3个抗纹枯病QTL位置与已报到的QTL位置均不一致,这些新发现的抗性QTL对进一步揭示纹枯病抗源CI12633的抗性机制提供帮助。
小麦纹枯病抗性QTL定位的关键是群体纹枯病抗性的鉴定,田间纹枯病抗性的鉴定受环境影响很大,气候条件、田间温湿度和其他小麦基部病害都会对纹枯病抗性鉴定造成干扰,本研究定位的QTL大多只在2~3个试验中出现,这也充分证明了这一点。因此,采用控温控湿等措施减少环境误差是今后进一步精确定位纹枯病抗性QTL的努力方向。另外,本研究遗传群体个体数偏小也一定程度影响QTL的检测,在今后的试验中将予以改进。
