CRISPR/Cas9系统靶向敲除多溴蛋白1基因对肝癌细胞功能的影响
2020-07-30宋金鸽焦宇辰
宋金鸽 焦宇辰
肝癌是全世界发病率最高的恶性肿瘤之一,其死亡率在肿瘤中位居第二位[1]。肝癌的肿瘤基因组研究已取得很大进展,TERT启动子、TP53、CTNNB1均为肝癌中的高频突变基因[2],而抑癌基因AT丰富结合域1A(ARID1A)作为染色质重塑的相关基因,在肝癌中也经常发生突变,且肝癌中ARID1A的表达量远低于癌旁组织[3]。但目前尚未发现其他染色质重塑相关基因对肝癌发生发展的作用。多溴蛋白1(PBRM1)基因编码ATP依赖的SWI/SNF染色质重塑复合物的亚单位,该亚单位控制DNA转录的可接近性[4]。既往研究表明,肝内胆管细胞癌中也存在17%的PBRM1基因突变[5]。在黄曲霉相关肝细胞肝癌中也发现8.2%的样本存在PBRM1基因突变[6]。因此,本研究通过应用规律成簇的间隔短回文重复(CRISPR)/CRISPR相关蛋白9(Cas9)系统基因编辑技术敲除人肝癌细胞系Huh7的PBRM1基因,构建PBRM1基因敲除的肝癌细胞模型,以进一步探讨PBRM1基因敲除后肝癌细胞的功能变化。
材料与方法
1.材料:人肝癌细胞株Huh7购自中国医学科学院基础医学研究所基础医学细胞中心;Huh7细胞、Huh7-KO细胞和HEK293T细胞均在添加10%胎牛血清、50 U/ml链霉素和青霉素的DMEM培养基中培养。
2.方法
(1)构建基因PBRM1双拷贝敲除的Huh7细胞株:从在线数据库(https://www.addgene.org/crispr/libraries/#geckov2)获得CRISPR-sgRNA序列,PBRM1-sgRNA上游引物:5’-CACCGAGGTTGTCGAATAACCA-3’,下游引物:5’-AAACTGGTTTTCCGACTCCTC-3’。EcoRI(美国,New England Biolabs公司)将LentiCRISPRv2(美国,Addgene公司)载体截切形成粘性末端,用T4DNA连接酶(美国,New England Biolabs公司)将剪切后的载体与退火后的sgRNAs通过粘性末端连接,得到一个稳定的表达载体。待HEK293细胞复苏后进行传代培养以确保细胞达到稳定状态,然后按每孔1×105个细胞传至六孔板中,培养1 d后,将表达载体、包装质粒pMD2.G(美国,Addgene公司)和psPAX2(美国,Addgene公司)共转染HEK293T细胞,培养48 h后收集包含病毒的细胞上清,以1 000 r/min离心10 min后保存于-80 ℃冰箱。病毒感染Huh7细胞48 h后用4 μg/ml嘌呤霉素筛选阳性克隆。为获得基因PBRM1双拷贝敲除克隆,将所选细胞置于96孔板上,每孔1个细胞,15 d后提取DNA,而后进行Sanger测序鉴定。PBRM1上游引物:5’-GTCTTCAGCCAGCCATA-3’,下游引物:5’-CCAAAGTGGAACCCAGTC-3’;扩增条件如下:95 ℃ 10 min共1个循环;94 ℃ 15 s,60 ℃ 30 s,72 ℃ 30 s,共35个循环;72 ℃ 5 min共1个循环。
(2)蛋白质印迹法(Western blot)检测PBRM1基因表达:用1%十二烷基硫酸钠处理细胞沉淀,然后在100 ℃下煮沸10 min,以1 2000 r/min离心10min至溶液澄清。布拉德福德蛋白检测试剂盒(中国,普利莱基因技术有限公司)用于蛋白质浓度定量。随后,在MES缓冲液(美国,Life Technologies公司)中将50~150 μg蛋白质装载于4%~12%的Bolt Bis Tris Plus凝胶(美国,Life Technologies公司)上。将蛋白质转移至0.45 μm硝化纤维素膜(美国,BioRad公司)中。在含5%脱脂奶粉的Tris缓冲盐水和0.1%吐温20(TBST)中,室温下将膜封闭1 h,然后在4 ℃下用一级抗体过夜培养。用TBST清洗膜,并在室温下用辣根过氧化物酶(HRP)结合的二级抗体孵育1 h。采用超敏化学发光法进行蛋白检测。应用Adobe Photoshop软件分析印迹。
(3)CCK-8法检测细胞增殖:以每孔5 000个细胞(100 μl细胞悬液)接种至96孔板,将96孔板置于37 ℃、5%CO2的培养箱中预培养。避光环境下,分别于24 h、48 h、72 h、96 h时间点向每孔加入10 μl的CCK-8溶液(日本,Dojindo公司),将96孔板在培养箱内放置4 h,使用酶标仪检测450 nm处的吸光度(OD值)。
(4)克隆形成实验:在96孔细胞板中按照每孔200个细胞进行铺板,每组设置3个复孔。将96孔板置于37 ℃、5%CO2的细胞培养箱中培养。定期查看,当培养皿中出现肉眼可见的克隆时,终止培养。,吸出并弃掉各孔的培养基,终止培养。用磷酸盐缓冲液(PBS)漂洗2次,4%多聚甲醛固定20 min,0.1%结晶紫(甲醇配制,PBS稀释)染色10 min,自来水冲洗后晾干,计算集落数(50个细胞以上)。采用酶联斑点图像自动分析仪进行拍照分析。
(5)细胞周期检测:10 μmol/L BrdU孵育细胞30 min,胰酶消化收集细胞后,用预冷的70%乙醇固定细胞,过夜。2 mol/L盐酸(含0.1 mg/ml胃蛋白酶)室温孵育20 min后PBS清洗,用50 μg/ml碘化丙啶(PI,美国,Sigma公司)和2 mg/μl 4’,6-二脒基-2-苯基吲哚(DAPI,美国,Sigma公司)染色。随后进行流式细胞仪检测。
(6)转录组测序技术(RNA-seq):分别取Huh7敲除(KO)细胞株和Huh7野生型(WT)细胞系的3组细胞,每组细胞分别提取RNA,进行后续操作。首先将提取后的RNA通过NanoDropTMOne/OneC检测纯度(OD260/280、OD260/230比值),经过Life Invitrogen Qubit©3.0荧光定量仪精确定量及Agilent 4200 TapeStation系统精确检测RNA完整性(RIN值)。随后进行文库构建,采用带Oligo(DT)的磁珠,对具有polyA结构的mRNA进行捕获。合成cDNA第1条链需要片段化mRNA(模板)、随机寡核苷酸(引物)及M-MuLV逆转录酶体系。合成cDNA第2条链需要先通过RNaseH降解RNA链,随后需要dNTPs(原料)和DNA Polymerase Ⅰ体系。将纯化后的cDNA进行末端修复、连接A尾和测序接头。使用AMPure XP beads筛选200 bp左右的cDNA,聚合酶链反应(PCR)扩增后需要再次使用AMPure XP beads纯化PCR产物,以获得最终的测序文库。质控合格后,按相应浓度和需求混合文库,随后进行Illumina X10测序仪PE150测序。待数据下机质控合格后,对两组样本的差异表达基因分别进行GO和KEGG功能富集,GO富集主要描述差异基因富集的生物过程(BP)、分子功能(MF)和细胞组成(CC)。KEGG富集分析主要查看差异基因所集中的信号通路。在GO和KEGG富集结果中选取校正P值(多重假设检验经过 BH[7]方法校正后的P值)小于0.05且富集基因数排名靠前的结果进行进一步讨论。
(7)γ-干扰素(IFN-γ)刺激实验:将Huh7-WT、Huh7-KO细胞分组,以每孔5 000个细胞(100 μl细胞悬液)接种至96孔板,将96孔板置于37 ℃、5%CO2的条件下预培养。6 h细胞贴壁后,吸出并弃掉培养基,按组加入含0 U/ml、10 U/ml、100 U/ml、1 000 U/ml IFN-γ的DMEM培养基,于培养箱中培养48 h。避光环境下,分别向每组的每孔加入10 μl的CCK-8溶液,将培养板置于37 ℃,5% CO2的条件中培养4 h,用酶标仪检测在450 nm处的OD值。计算抑制率,抑制率=[(Ac-As)/(Ac-Ab)]×100%[As:实验孔(含有培养基、CCK-8、IFN-γ);Ac:对照孔(含有培养基、CCK-8,不含IFN-γ);Ab:空白孔(不含细胞和IFN-γ的培养基、CCK-8)]。

结 果
1.PBRM1双拷贝敲除Huh7细胞株的构建:Sanger测序显示目的区域被成功敲除(图1A),表明设计的sgRNA可高效编辑PBRM1基因。Western blot检测转染后的单克隆细胞株Huh7-KO,结果显示Huh7-KO细胞株中PBRM1蛋白表达缺失,而Huh7-WT细胞系PBRM1蛋白表达正常,内参GAPDH表达量正常且基本一致(图1B),说明本研究在Huh7细胞系中成功敲除了PBRM1基因。
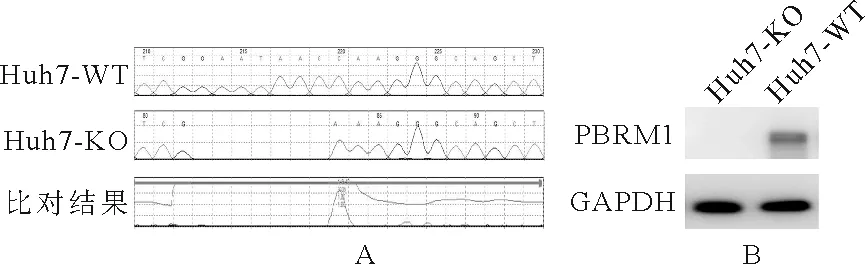
图1 PBRM1基因敲除细胞株的成功构建[A:Huh7-KO细胞株与Huh7-WT细胞系的DNA在PBRM1基因目的区域的Sanger测序峰图比对;B:Western blot检测Huh7-KO细胞株和Huh7-WT细胞系的PBRM1蛋白表达)
2.PBRM1基因的敲除抑制肝癌细胞增殖:培养48 h后,与Huh7-WT细胞系比较,Huh7-KO细胞株增殖活性明显受到抑制(1.254±0.035比0.843±0.029,P<0.000 1);72 h后抑制效果更加显著(1.858±0.050比0.945±0.028,P<0.000 1,图2A)。克隆形成实验结果显示,与Huh7-WT细胞系比较,Huh7-KO细胞株的克隆形成数偏低,克隆形成明显受到抑制(图2B)。

图2 Huh7-KO细胞株的细胞增殖情况[A:CCK-8测定的OD值96 h内在Huh7-KO细胞株和Huh7-WT细胞系中的变化(P<0.000 1);B:平板克隆形成实验结果]
3.表达差异基因的功能富集:Huh7-KO细胞株与Huh7-WT细胞系相比共筛选出5 603个差异表达基因,其中3 303个基因表达上调,2 300个基因下调。GO富集结果显示,表达下调的相关BP主要关于细胞周期,如DNA复制(P<0.000 1,富集基因数=90)、染色体分离(P<0.000 1,富集基因数=100)、核分裂(P<0.000 1,富集基因数=96)、细胞时相过渡的下调(P<0.000 1,富集基因数=98)、核糖体的合成(P<0.000 1,富集基因数=74);表达下调的CC主要与细胞分裂有关,包括中心体(P<0.000 1,富集基因数=83)、染色体(P<0.000 1,基因富集数=100)、纺锤丝(P<0.000 1,基因富集数=77)等;MF出现下调的通路主要包括核糖体结构组成(P<0.000 1,富集基因数=67)、微管蛋白的结合(P<0.000 1,富集基因数=56)等(图3A);而表达上调的BP包括适应性免疫应答(P<0.000 1,富集基因数=86)及细胞对IFN-γ的反应(P<0.000 1,富集基因数=45)。KEGG富集结果显示,细胞周期(P<0.000 1,基因富集数=46)、DNA复制(P<0.000 1,富集基因数=28)及核糖体相关通路(P<0.000 1,富集基因数=56)明显下调;而JAK-STAT相关基因表达上调(P=0.028,基因富集数=38)。
4.PBRM1基因的敲除使Huh7肝癌细胞出现G1/S阻滞:Huh7-WT细胞系中处于G1期细胞的平均比例为(57.23±1.71)%,而Huh7-KO细胞株为(74.9±1.61)%,明显高于Huh7-WT细胞系(P=0.016 6)。Huh7-WT细胞系中处于S期细胞的平均比例为(36.5±1.36)%,而Huh7-KO细胞株为(19.8±1.58)%,明显低于Huh7-WT细胞系(P=0.000 2)。Huh7-WT细胞系和Huh7-KO细胞株处于G2/M期细胞的平均占比分别为(6.27±0.37)%和(5.26±0.59)%,差异无统计学意义(P>0.05)。见图3。
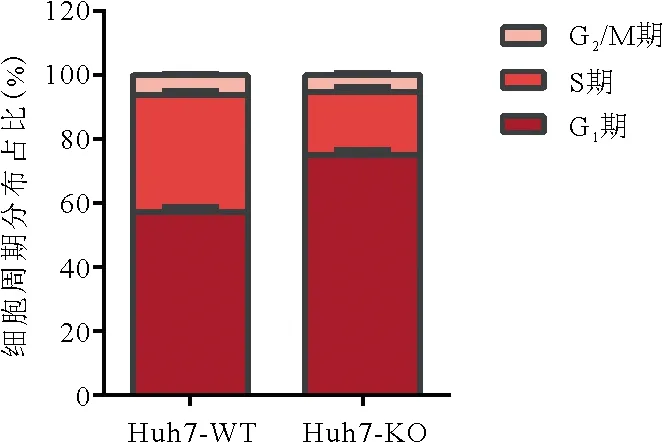
图3 Huh7-WT细胞系和Huh7-KO细胞株在细胞周期中各时期细胞所占比例
5.PBRM1敲除可增强Huh7细胞对IFN-γ治疗的敏感性:与此同时,在GO通路富集中,表达上调的BP还包括细胞对IFN-γ的反应(P<0.000 1)。并且在KEGG分析中,我们发现上调基因富集于JAK-STAT通路,而该通路为IFN-γ介导的信号通路[8],因此将Huh7-WT细胞系和Huh7-KO细胞株分别在不同浓度梯度的IFN-γ中进行培养,验证GO和KEGG功能富集的结果。结果显示,1 000 U/ml IFN-γ对Huh7-KO细胞株组的细胞抑制作用明显高于Huh7-WT细胞系组[抑制率分别为(58.32±3.28)%和(48.67±1.32)%,P=0.009 1];未加IFN-γ的Huh-WT细胞系组的增殖率明显高于Huh7-KO组[增殖率分别为(23.53±4.75)%和(45.88±5.46)%,P=0.005 9]。见图4。
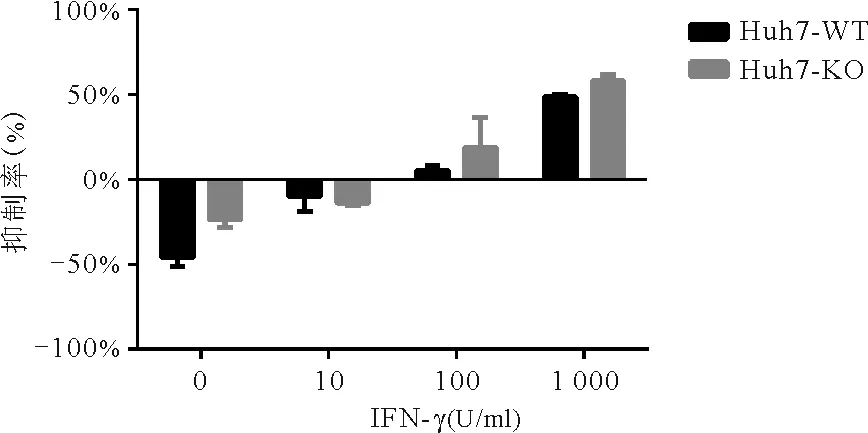
图4 不同浓度IFN-γ对Huh7-WT和Huh7-KO的抑制作用(y轴中负值表示细胞生长未受到抑制,依旧处于增殖阶段,对应数值为增殖率)
讨 论
本研究首次在肝癌细胞株中敲除PBRM1基因,并发现该基因的敲除可以导致细胞G1/S期阻滞从而抑制细胞株增殖。而PBRM1基因通常作为抑癌基因参与染色质重塑在肾透明细胞癌中具有较高的突变频率[9-10],有研究报道,约40%的肾透明细胞癌患者存在PBRM1基因突变[11],且PBRM1和BAP1的缺失与患者较差的预后相关[12]。在肾透明细胞癌的治疗方面,PBRM1基因的功能缺失性突变与程序性死亡受体1(PD-1)治疗有效显著相关[13]。在肾透明细胞癌中进行PBRM1基因的敲除可以导致80%肾癌细胞增殖显著增加[11],与本研究的结果相反。这可能由于PBRM1基因在不同肿瘤中的功能存在差异。且PBRM1基因的表达量和预后关系在不同类型肿瘤中也不尽相同,在肾透明细胞癌、结肠癌中PBRM1基因表达量较低的患者预后较差[14-15],但在子宫内膜癌中,PBRM1表达量的阳性率随病理分级和临床分期的增高而增加[16]。从网站The Human Protein Atlas中的PBRM1蛋白免疫组化染色结果也可以发现,肝癌中PBRM1基因的表达量高于正常肝脏组织。而PBRM1基因的表达量与肝癌的预后相关性研究尚有待开展。
众所周知,肝脏切除和肝脏移植是早期肝癌的主要治疗手段,对于不适合肝脏切除、肝脏移植或局部治疗的患者,主要选择索拉菲尼和乐伐替尼进行多靶点分子治疗[17]。即使这些治疗可以提高患者生存期,但较大程度的细胞抑制和治疗耐药也是患者长期生存的重大限制。因此,有研究对晚期肝癌患者采用免疫检查点阻断治疗。曲美母单抗是首次用于晚期丙型肝炎病毒(HCV)相关的肝细胞肝癌患者的细胞毒性T淋巴细胞相关蛋白4(CTLA-4)单抗,其治疗应答率适中(17%),应答持续中位时间为6.5个月[18]。也有在亚洲地区进行的临床试验采用PD-1抗体纳武利尤单抗治疗索拉菲尼经治的晚期肝癌患者,客观应答率为15%[19]。本研究结果显示,敲除PBRM1基因的细胞株中表达上调的基因主要富集于免疫应答,提示PBRM1基因敲除可能有助于免疫系统的活化,免疫治疗的有效性可能会由于PBRM1的缺失得到进一步提升。在其他类型肿瘤中,也有证据表明PBRM1缺失与免疫治疗获益相关。PBRM1基因中的功能缺失性突变使黑色素瘤模型小鼠对免疫检查点阻断治疗的联合用药反应更加敏感,若无该突变,则会产生对免疫治疗的耐药[20]。在肾透明细胞癌中也发现,PBRM1缺失与免疫检查点阻断治疗的临床获益相关[13]。
本研究还发现,Huh7-KO细胞株对IFN-γ的抑制作用更敏感,且IFN-γ介导的JAK-STAT通路相关基因表达上调。曾有研究表明JAK2的缺失会使PD-L1及主要组织相容性复合体Ⅰ类分子(MHC-Ⅰ)表达下调从而产生免疫阻断治疗耐受[21],本研究的RNA-seq结果显示,Huh7-KO细胞株JAK2表达上调,提示PBRM1基因的敲除可能通过促进免疫应答从而影响免疫检查点阻断对肝癌治疗的疗效。IFN-γ的体外治疗可通过与受体结合并激活JAK-STAT信号通路,直接抑制肿瘤细胞生长并促进肿瘤细胞凋亡[22]。随后有研究表明,在非小细胞肺癌中,不同浓度IFN-γ对肿瘤细胞的作用不尽相同,低浓度IFN-γ优先激活ICAM1-磷酸肌醇3激酶/蛋白激酶B(PI3K-Akt)-Notch1通路诱导肿瘤的发展和转移,高浓度IFN-γ优先激活JAK-STAT通路,诱导肿瘤细胞凋亡[23]。我们通过IFN-γ刺激实验也发现低浓度IFN-γ促进肿瘤细胞增殖、高浓度抑制增殖的现象。同时,IFN-γ信号通路还可以在抗肿瘤免疫中发挥免疫调控作用,可能会使免疫治疗产生原发性耐药和获得性耐药[24-25]。IFN-γ信号通路的功能丧失会使肿瘤对CTLA4阻断治疗产生原发性耐药导致治疗无效。而长期的IFN-γ暴露会使肿瘤细胞产生免疫逃逸,对免疫检查点治疗产生获得性耐受,通过JAK抑制剂关闭干扰素途径,可以改善免疫检查点阻断治疗的耐受问题[24]。还有研究表明,RIG-I表达量高的肝癌患者对于IFN-α治疗更加有效[26]。但是在肝癌细胞株中PBRM1基因的敲除与免疫治疗的相关性还有待探索。
综上所述,在人肝癌细胞系Huh7中敲除PBRM1基因可以通过G1/S期阻滞抑制细胞增殖,且IFN-γ的杀伤作用在敲除PBRM1基因后的Huh7细胞株中更加显著。本研究结果为进一步探讨PBRM1基因在肝癌中的功能和作用奠定基础,为肝癌的治疗提供了新的思路。
