羰基铁粉掺杂整体柱萃取-在线热解吸进样气相色谱-串联质谱法分析茶叶样品中拟除虫菊酯类农药残留
2020-07-27吴永慧吕亚宁淦五二
吴永慧, 邓 云, 吕亚宁, 淦五二*
(1. 中国科学技术大学化学系, 安徽 合肥 230026; 2. 合肥海关技术中心, 安徽 合肥 230022)
固相微萃取(SPME)是一种简单、高效且无溶剂的样品制备技术,可用于选择性吸附和富集样品溶液中的目标分析物[1,2],主要包括光纤SPME[3-5],管内SPME[6,7]和薄膜SPME[8,9]等模式。但它们的萃取层薄且固定相体积小,吸附容量和测定灵敏度并不令人满意[10]。Baltussen等[11]提出了搅拌棒吸附萃取(SBSE)技术,提高了方法吸附容量和富集倍数[12]。但由于吸附和热解吸过程分离,难于进行目标分析物在线吸附/解吸过程。
拟除虫菊酯是一类重要的合成杀虫剂[13,14]。与传统的有机氯农药相比,它们具有高效、低毒且可生物降解的特性[15],近年来已被广泛用于预防和控制茶产品中的害虫。但使用不当会导致较高残留量,严重威胁人类健康,直接影响茶叶产品出口。常用检测茶叶中拟除虫菊酯类农药的残留方法是气相色谱与电子捕获检测器联用(GC-ECD)[16,17],与火焰离子化检测器联用(GC-FID)或与质谱检测器联用(GC-MS)[18-20]。由于茶叶中农药残留含量低,测定前需要进行预富集,但传统的液液萃取和固相微萃取富集倍数往往不能令人满意,且不能实现与测量仪器在线联用。
本文拟建立一种可以实现整体柱富集和在线热解吸方法,并与GC-MS/MS联用检测茶叶中拟除虫菊酯类农药残留。目标分析物在样品流过整体柱时被吸附和浓缩。随后,将整体柱置于高频电磁感应加热装置中,实现目标分析物在线热解吸进样。本工作提高了方法的富集倍数,降低了检出限,可以应用于实际样品中相关农药残留成分的分析检测。
1 实验部分
1.1 仪器、试剂与材料
GCMS-TQ8040系统气相色谱-三重四极杆质谱仪由GC-2010 Plus气相色谱仪和TQ-8040三重四极杆质谱仪组成(岛津); Rtr-5MS毛细管柱(30 m×0.25 mm×0.25 μm)(岛津); AX-650扫描电子显微镜(SEM)(日立); H-7650透射电子显微镜(TEM)(日立); EQUINX红外光谱仪(布鲁克); DTG-60H热重分析仪(岛津); UV-VIS-756分光光度计(托普分析仪器有限公司); IFIS-C蠕动泵(瑞迈电子科技有限公司)。
羰基铁粉(内径6 μm)购自巴斯夫。端羟基聚二甲基硅氧烷(PDMS-OH)(550 g/mol)购自西格玛。四乙氧基硅烷(TEOS)、聚乙二醇(PEG)、乙醇、二乙胺购自国药集团化学试剂有限公司。
氯菊酯(permethrin,纯度99%)、氟氯氰菊酯(cyfluthrin,纯度98%)、氯氰菊酯(cypermethrin,纯度98%)、α-氯氰菊酯(alphacypermethrin,纯度99%)和溴氰菊酯(deltamethrin,纯度99%)购自阿尔塔科技有限公司。
茶叶样品:黄山毛峰(绿茶)和祁门红茶(红茶),购自当地市场。
萃取/解吸系统示意图如图1所示:目标分析物富集于自制的整体柱中,阀门1、2用于切换载气净化模式并收集废液。自制电磁感应加热线圈(EIHC)缠绕在整体柱上,并连接到高频感应加热电源(HFPS)上进行热解吸过程。
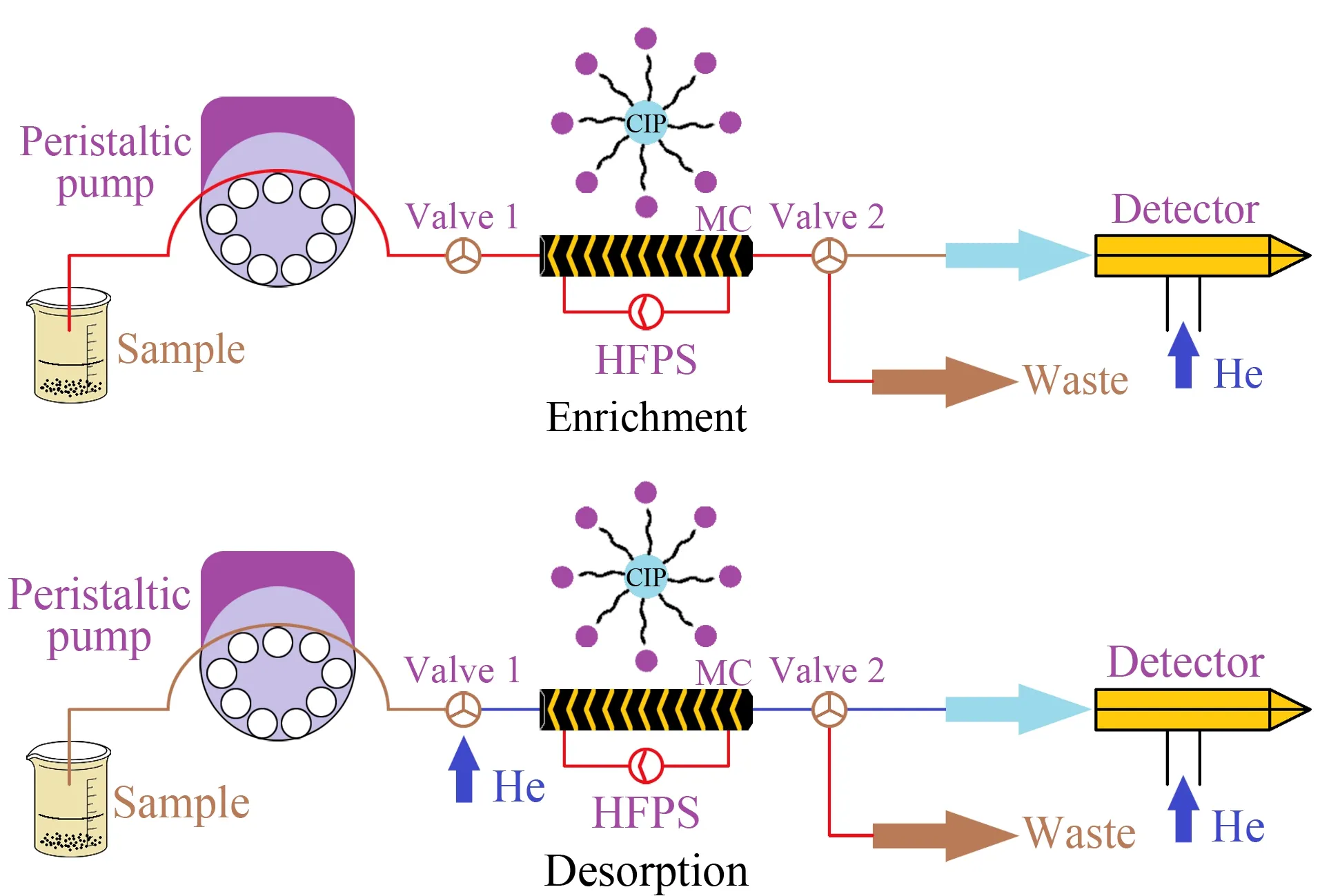
图 1 萃取/解吸系统示意图Fig. 1 Schematic of extraction/desorption system HFPS: high-frequency induction heating power source; MC: monolithic column.
1.2 实验条件
气相色谱条件 温度程序:柱初始温度为50 ℃(保持1 min),然后以25 ℃/min的速度升至125 ℃,最后以10 ℃/min的速度升至300 ℃并保持15 min;载气为He,柱流速为1.69 mL/min;使用不分流进样模式;入口温度:250 ℃,恒定线速度模式(47.2 cm/s); GC-MS接口温度为250 ℃。
MS/MS条件 离子源:电子轰击(EI), 70 eV;离子源温度:200 ℃;检测电压:0.6 kV;喷油器温度:250 ℃;溶剂延迟时间:1.5 min;多反应监测(MRM)模式。
热解吸条件 解吸温度250 ℃;解吸时间100 s;载气流速30 mL/min。
于0.7 mL 0.6 mol/L HCl溶液中逐滴加入1.5 mL TEOS和2 mL乙醇,搅拌至溶液澄清透明后,加入1.5 mL PDMS-OH, 60 ℃下恒温1 h。缓慢滴加0.5 mL 15%(v/v)二乙胺乙醇溶液,并在激烈搅拌下加入2.50 g羰基铁粉(carbonyl iron powder, CIP)。将所得悬浮液转移至一端密封的熔融石英管中后将另一端密封,于60 ℃下恒温12 h。最后,分别用乙醇和超纯水将整体柱洗涤3次,并在85 ℃下干燥24 h。
将整体柱置于石英管中,在氮气保护下进行老化:室温→120 ℃(保持3 h)→240 ℃(保持3 h)→280 ℃(保持4 h)。升温速率均为1 ℃/min。最后,用水和乙醇清洗整体柱若干次,并置于60 ℃下干燥备用。
1.4 标准溶液和茶叶样品制备
配制200 mg/L拟除虫菊酯甲醇标准储备溶液,用10%甲醇溶液逐级稀释得到标准工作溶液。茶叶样品粉碎并通过40目筛,放入密封袋中使用。称取15.000 g茶叶样品(或空白样品)于三角瓶中,加入30 mL 10%甲醇溶液,超声提取6 h,然后使用整体柱对样品进行纯化以备后续使用。
1.5 分析过程
取2 mL样品溶液于20 mL样品瓶中,加入18 mL蒸馏水。将样品溶液以2.0 mL/min的流速通过整体柱后,旋转阀1,载气(He, 30 mL/min)吹扫残留的样品溶液15 s。关闭阀1,打开HFPS,持续100 s。最后关闭阀1、阀2,目标分析物由载气引入气相色谱仪进行GC-MS/MS分析。离子通道1用于定量分析,其他离子通道用于定性分析。相关数据见表1。运行时间36.50 min。

表 1 5种拟除虫菊酯类农药目标物的离子通道数据
2 结果与讨论
2.1 整体柱的制备
2.1.1磁性颗粒的选择
整体柱制备过程如图2所示,TEOS水解产物脱水形成SiO2无机网络,PDMS-OH通过共价键与其聚合。CIP加入到溶胶溶液中后,CIP表面羟基与TEOS水解产物残留羟基聚合。
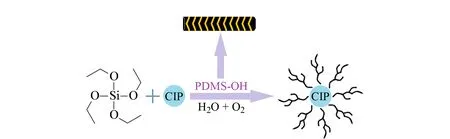
图 2 整体柱材料制备过程示意图Fig. 2 Schematic diagram of the preparation process of monolithic column material

图 3 不同磁性颗粒整体柱的扫描电镜图Fig. 3 Scan electron microscope(SEM) images of materials with magnetic particlesa. CIP; b. reduced iron powder.
磁性颗粒在整体柱中充当加热介质的角色,其分散均匀性直接影响样品的热解吸效率。研究分别采用还原铁粉(30 μm i. d.)和CIP磁性颗粒合成整体柱,扫描电镜图像如图3所示。可以看出,还原铁粉和CIP磁性颗粒均形成了有机-无机杂化材料骨架结构,但含有CIP磁性颗粒的整体柱骨架结构更明显且分布均匀,且由于CIP磁性颗粒粒径较均匀,具有高活性,在高频磁场下拥有高磁通率,并可通过铁氧键结合成核,通过Si-O-Si键聚集生长,形成包裹层[21],因此实验选用CIP磁性颗粒作为整体柱填充材料。
2.1.2溶剂的选择及用量
实验使用摩尔质量为550 g/mol的PDMS-OH,它可以很好地溶于乙醇中。PDMS-OH在整体柱制备中具有两种作用,一种是增强杂化材料的韧性,TEOS/PDMS体积比通常为5/5至7/3[22],随着体积比的增加,整体柱材料收缩和断裂概率也增大;但当体积比低于5/5时,也会导致柱材料内部网络结构发生断裂。同时,PDMS-OH还作为整体柱的固定相,有机成分含量增加有助于提高材料的吸附性能。研究表明当TEOS和PDMS的体积比为3/2时,整体柱具有良好的网络结构和吸附性能。
PEG常用作整体柱制备过程中的致孔剂[23]。适量的PEG不仅可以保证良好的孔隙率,还可以保证骨架强度。实验研究发现,当PEG为0.2 g时,所得整体柱的孔隙率大于85%,具有良好的渗透性和稳定的骨架结构。
2.1.3酸/碱催化剂
酸和碱均可用作制备整体柱的催化剂。考虑到在酸性和碱性条件下水解和缩合的相对反应速率差异,本工作中使用两步酸/碱催化法。
盐酸选为TEOS水解催化剂。实验发现酸浓度会影响水解时间,当HCl浓度从0.1 mol/L变化到0.8 mol/L时,水解时间即从4 h减少到1 h。当浓度大于0.8 mol/L时,水解时间将进一步缩短。
二乙胺乙醇溶液为缩合催化剂。实验发现当二乙胺溶液的体积分数达到15%(v/v)以上时会出现白色沉淀。当二乙胺溶液的体积分数从15%降至14%时,胶凝时间即从30 s延长至3 min,导致CIP聚沉。基于水解和形成凝胶的时间范围,本实验采用0.6 mol/L HCl和15%二乙胺乙醇溶液。得到的凝胶pH约为7。
2.1.4含水量
含水量是网络形成的关键因素。含水量减少将减缓TEOS的水解速率,进而导致TEOS和PDMS之间的缩合进程减缓。然而,水含量过高会发生自缩合作用,出现材料结块情况。图4显示了在不同含水量下整体柱材料的SEM图像。结果表明,含水量较低时,呈现出许多大小不同的球形颗粒,但未形成网络骨架结构(0.6 mL,图4a)。而含水量过多时,材料发生了聚结(0.8 mL,图4c)。当使用0.7 mL水时,材料形成了清晰的网络结构(图4b)。因此,本实验使用的水含量为0.7 mL,也即加入的HCl体积为0.7 mL。

图 4 不同含水量下材料的SEM图像Fig. 4 SEM images of materials under different water contentsa. 0.6 mL; b. 0.7 mL; c. 0.8 mL.
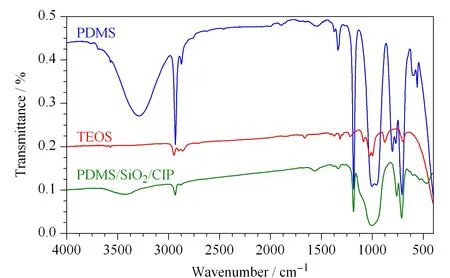
图 5 整体柱材料的红外光谱图Fig. 5 Infrared spectra of PDMS, TEOS and CIP/SiO2/PDMS
2.2 羰基铁粉掺杂整体柱(CIP/SiO2/PDMS)杂化材料的特性
研究了整体柱的红外光谱和热重特性。从图5可知PDMS和CIP/SiO2/PDMS在3 308 cm-1附近的吸收带是羟基的不对称拉伸振动。缩合后,吸收强度变弱,这意味着许多羟基已经反应,Si-OH吸收带消失同时PDMS也已连接到无机SiO2网络上。2 964 cm-1附近的强峰为-CH3,这些峰在水解后仍保留,表明PDMS已经连接到CIP表面。Fe-O-Si的特征吸收峰位于633 cm-1和561 cm-1,表明杂化材料已经通过Fe-O-Si化学键连接到CIP的表面。
整体柱热重特性显示失重分为3个阶段(见图6):第一阶段,低于300 ℃时失重是源于水的蒸发以及残留在材料中的有机试剂挥发和降解;第二阶段,在300 ℃至420 ℃之间的失重可能是填充剂等聚合小分子降解;第三阶段,420 ℃后,失重急剧增加,表明在这个温度下,材料开始迅速分解。因此,整体柱在300 ℃以下具有热稳定性,可以满足工作要求。
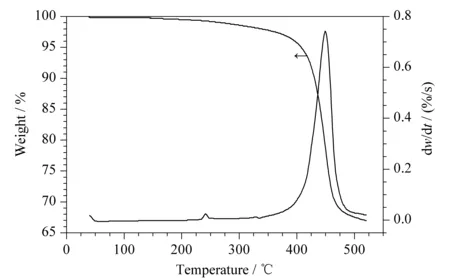
图 6 CIP/SiO2/PDMS杂化材料的热重曲线Fig. 6 TGA curve of the CIP/SiO2/PDMS material
2.3 样品预处理条件的优化
研究了丙酮、乙腈和甲醇3种常用提取溶剂对茶叶的提取效果。结果表明,甲醇对目标分析物的总提取效率最高,其次是乙腈和丙酮。因此,本实验选择甲醇溶液作为提取溶剂。
研究了提取时间以及氯化钠含量对提取效率的影响。结果表明,提取效率随提取时间延长而增加,6 h后趋于平稳。因此,选择6 h萃取时间以确保达到平衡。而盐析效应不会明显提高目标分析物的提取效率。相反,随着NaCl含量的增加,目标分析物的提取量反而减少,因此,提取时不在样品中添加氯化钠。
2.4 整体柱萃取条件的优化
2.4.1整体柱的饱和吸附容量
配制0.3 mg/mL的拟除虫菊酯目标物溶液,使其流过整体柱,然后用紫外吸收光谱法进行在线检测。综合考虑柱压力和分析速度,选择溶液流速为2 mL/min。根据上样溶液的质量浓度、流速和时间计算饱和吸附容量,5种拟除虫菊酯的吸附容量分别为18 mg(氯菊酯)、25 mg(氟氯氢菊酯)、31 mg(氯氰菊酯)、29 mg(α-氯氰菊酯)和45 mg(溴氰菊酯)。
2.4.2提取溶剂甲醇的含量和柱流速
样品提取物中甲醇的体积分数会影响整体柱的富集效率。研究结果表明,目标分析物的富集效率开始随甲醇体积分数增加而增加,甲醇体积分数高于10%后富集效率下降。因此,本实验中选择甲醇的体积分数为10%。
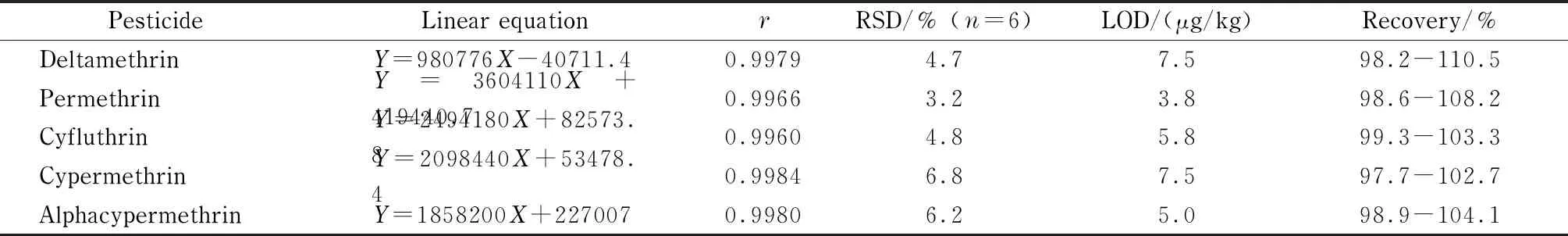
表 2 5种农药的线性关系、峰面积的RSD、检出限和回收率
研究了样品流速(1.0~3.0 mL/min)对目标分析物富集效率的影响,结果如图7所示,样品流速设置为2.0 mL/min时,可获得最佳的实验结果。
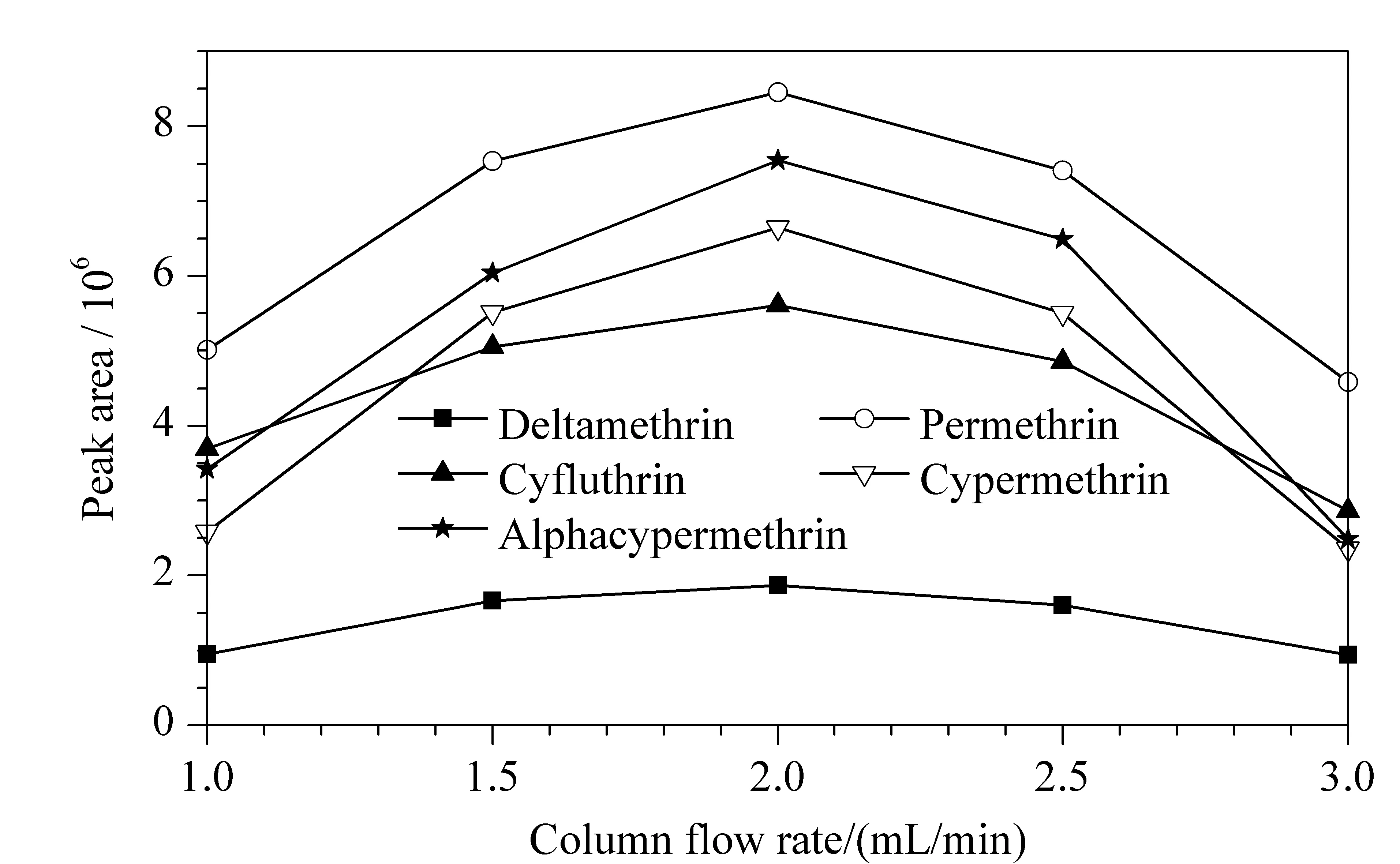
图 7 不同柱流速对5种拟除虫菊酯类农药目标物的富集效率Fig. 7 Enrichment efficiency of five pyrethroid pesticide targets with different column flow rates
2.5 解吸条件优化
2.5.1解吸温度
不同高频感应加热功率下温度-时间变化情况如图8所示,当功率为200 W时,10 s内温度可达到250 ℃。为进行有效的样品热解吸,加热温度应高于其沸点(170~195 ℃),并低于其分解温度和整体柱承受温度(300 ℃)。因此,工作温度设定为250 ℃,相应的电磁感应加热功率为200 W。
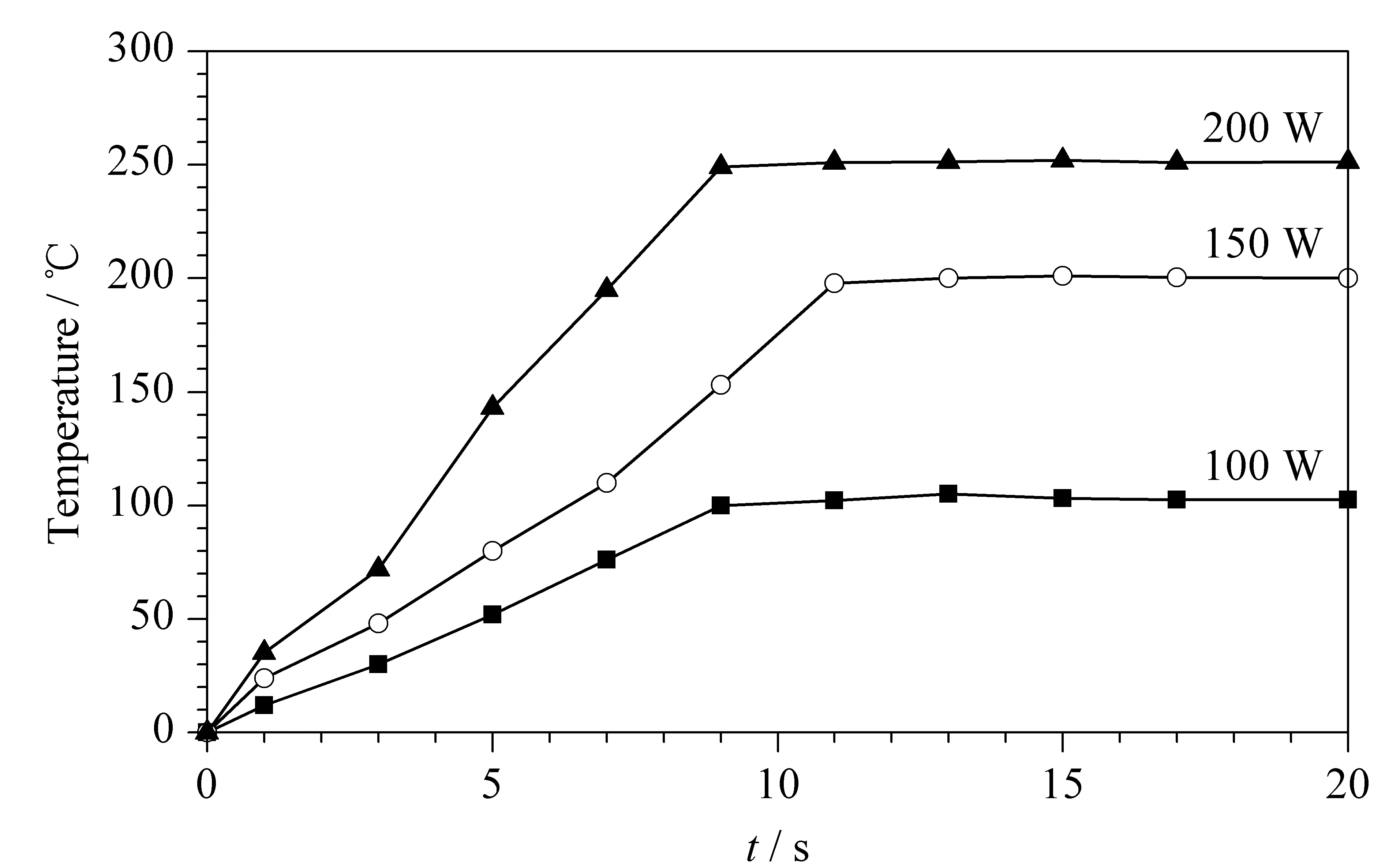
图 8 整体柱在不同功率下的温度-时间曲线Fig. 8 Curve of temperature-time of the monolithic column under different power
2.5.2解吸时间
实验研究了在20 s至180 s范围内不同解吸时间下拟除虫菊酯类农药的色谱峰面积,结果表明峰面积在100 s处峰值达到最大,随后不再增加。因此,实验中解吸时间设置为100 s。
2.5.3载气流速
载气流速太低,会导致目标分析物难以快速进样,从而使得峰形变宽且容易产生重叠。相反,如果载气流速太快,柱压力则会变高,保留时间提前,得到的峰形往往比较尖锐。当载气流速设置为30 mL/min时,可获得最佳结果。故实验中的载气流速设定为30 mL/min。
2.6 方法评估
将20 mL 0.2、0.4、0.6、0.8、1.0 mg/L标准溶液和样品溶液以2.0 mL/min的流速依次通过整体柱进行萃取富集,然后进行分析,记录峰面积,获得每种成分的标准曲线(峰面积和质量浓度之间的线性关系);取0.6 mg/L标准溶液,连续进样6次,测定峰面积的RSD,结果见表2。根据信噪比为3(S/N=3)确定检出限(LOD)。在优化后的实验条件下,分别在绿茶、红茶空白样品中做加标回收试验,添加水平为0.02、0.1和0.5 mg/kg,每个水平做6个平行样,得到的检出限和回收率数据列于表2,结果显示本方法具有良好的分析性能。
采用常规直接进样法测定了50 μg/L的拟除虫菊酯类农药溶液,同时用本方法测定0.05 μg/L拟除虫菊酯溶液,研究方法的富集倍数。从图9可以看出,两种方法峰面积相似,表明在最佳条件下,本方法的富集倍数约1 000倍。
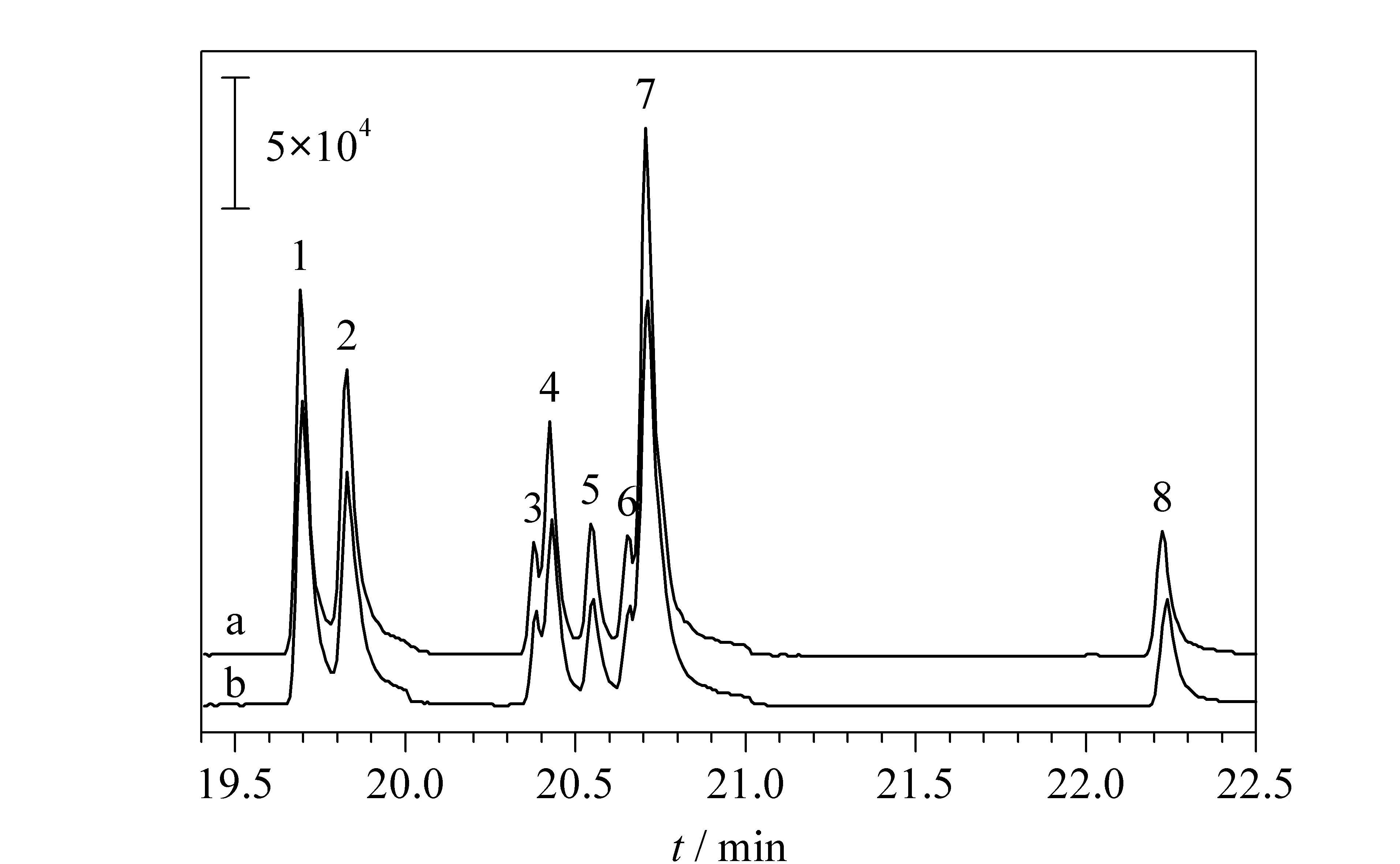
图 9 (a)常规方法和(b)本方法的比较Fig. 9 Comparison of (a) conventional method and (b) this method 1. permethrin 1; 2. permethrin 2; 3. cyfluthrin 1; 4. cyfluthrin 2; 5. cypermethrin 1; 6. cypermethrin 2; 7. alphacypermethrin; 8. deltamethrin 2.
2.7 实际样品分析
测定了市场上随机购买的6个茶叶样品,在1个茶叶样品中检出了氯菊酯(含量为0.91 μg/kg)、氯氰菊酯(含量为6.5 μg/kg)和α-氯氰菊酯(含量为13.8 μg/kg),在1个茶叶样品中检出氯菊酯(含量为0.89 μg/kg)、氯氰菊酯(含量为7.03 μg/kg)、α-氯氰菊酯(含量为12.1 μg/kg)和溴氰菊酯(含量为15.0 μg/kg),但含量均没超过欧盟最高残留量(MRL)规定(氯氰菊酯的MRL低于0.5 mg/kg、氯菊酯的低于0.1 mg/kg)。其他4个样品中未检出拟除虫菊酯类农药残留。
3 结论
本文制备了羰基铁粉掺杂硅胶整体柱,用于拟除虫菊酯类农药残留萃取,并与GC-MS/MS法联用,建立了在线富集、热解吸GC-MS/MS测定茶叶样品中拟除虫菊酯类农药残留方法。首先,目标分析物吸附并浓缩在整体柱中;其次,在高频电磁感应加热装置中,目标分析物实现了热脱附。本方法简便,耗时短,灵敏度高,重现性好,回收率稳定,能够满足国内外对茶叶中多种拟除虫菊酯类农药残留的检测要求。整体柱与GC-MS/MS的联用也为样品前处理及复杂基质的富集分析检测提供了新思路。
