QuEChERS-高效液相色谱法同时测定植物固体饮料中10种植物毒素
2020-07-26张鹏斐
张鹏斐
(湖南省怀化市食品药品检验所,湖南 怀化 418000)
植物固体饮料是以植物及其提取物为主要原料加工制成的一类冲泡型饮料,近些年被作为代餐减肥食品受到了很多人青睐,然而其安全隐患也随之而来。从20世纪90年代“植物毒素”[1-2]这一名称的提出到最近几年关于植物类食品安全隐患的研究报道[3-5],均表明相关学者对植物类食品的安全性提出了质疑。并且,欧盟、澳大利亚和新西兰等国家和地区对饮料中10种植物毒素(芦荟苷、β-细辛脑、黄连素、黄樟素和异黄樟素、山道年、原百部碱、长叶薄荷酮、侧柏酮、香豆素)的含量进行了严苛规定[6],并以此管控中国出口产品,而中国相关标准与法规还相对缺乏[7]。
目前,植物毒素的样品处理方法仅有传统的固相萃取法[8-9],且均用于液体饮料中,并不适用于固体饮料,而且用该方法处理样品后提取液浑浊不清不适用于检测。相较于上述方法,QuEChERS法操作简单、吸附性高、效率高、提取液澄清[10-12],更适用于成分复杂的植物固体饮料成分提取。关于植物毒素的检测方法主要有气相色谱法[13]、气相色谱—质谱法[14-15]、液相色谱法[16-17]及液相色谱—串联质谱法[18]等,其中,高效液相色谱法为主要方法,对于复杂组分分析有明显优势,且DAD检测器能通过DAD图谱比对判断检测成分是否为所测成分。综上考虑,试验拟采用QuEChERS方法对市售的植物固体饮料进行提取、净化,用超高效液相色谱DAD定性定量,以建立同时检测植物固体饮料中10种植物毒素的方法,以期为植物固体饮料的日常监督提供有利且客观的技术依据。
1 试验方法
1.1 仪器与设备
高效液相色谱仪:e2695型,沃特世科技(上海)有限公司;
高速冷冻离心机:ST 16R型,赛默飞世尔科技(中国)有限公司;
电子天平:AE2204型,湘仪天平仪器设备有限公司;
超声清洗机:Model 9960B型,天津科贝尔光电技术有限责任公司;
可调式混匀仪:MX-S型,大龙兴创实验仪器(北京)有限公司;
超纯水仪:Synergy High flow型,默克化工技术(上海)有限公司。
1.2 试剂与材料
原百部碱(CAS:27495-40-5):纯度99.67%,德国Dr.Ehrenstorfer GmbH公司;
香豆素(CAS:91-64-5):纯度100%,德国Dr.Ehrenstorfer GmbH公司;
黄连素(CAS:633-65-8):纯度97.15%,德国Dr.Ehrenstorfer GmbH公司;
山道年(CAS:481-06-1):纯度99.80%,德国Dr.Ehrenstorfer GmbH公司;
芦荟苷(CAS:1415-73-2):纯度99.11%,德国Dr.Ehrenstorfer GmbH公司;
侧柏酮(CAS:1125-12-8):纯度98.4%,德国Dr.Ehrenstorfer GmbH公司;
β-细辛脑(CAS:5273-86-9):纯度98.06%,德国Dr.Ehrenstorfer GmbH公司;
长叶薄荷酮(CAS:89-82-7):纯度99.8%, 中国食品药品检定研究院;
黄樟素(CAS:94-59-7):纯度99.6%,美国Stanfordchem公司;
异黄樟素(CAS:120-58-1):纯度97.0%,美国AccuStandard公司;
乙腈、甲醇:色谱纯,美国GRACE公司;
乙酸铵:色谱纯,美国ACS公司;
甲酸:色谱纯,天津科密欧化学试剂有限公司;
氯化钠、无水硫酸镁、醋酸钠:分析纯,国药集团化学试剂有限公司;
十八烷基硅烷键合硅胶(C18)、乙二胺-N-丙基硅烷化硅胶(PSA):40~60 μm,武汉市伟琪博星生物科技有限公司;
石墨化碳黑(GCB):40~120 μm,武汉市伟琪博星生物科技有限公司;
分析样品(10种植物固体饮料):市售。
1.3 标准溶液配制
1.3.1 植物毒素标准储备液的配制 将10种植物毒素分别用甲醇配制成约1.0 g/L的标准浓度的储备溶液,将其放置在4 ℃的冰箱里避光冷藏,可储存6个月。
1.3.2 植物毒素混合标准溶液的配制 分别吸取上述适量的10种植物毒素的单标储备液,用30%甲醇—70% 10 mmol/L乙酸铵溶液(pH 3.5)稀释成浓度约为200 mg/L的混合标准溶液。测试时,用30%甲醇—70% 10 mmol/L乙酸铵溶液(pH 3.5)将混合标逐步稀释成用于检测灵敏度和线性范围分析配制的标准工作溶液。
1.4 色谱条件的探索
之前有研究者[8]发现有些植物毒素更具碱性和极性。进行HPLC检测时,容易出现色谱峰形状不佳,拖尾和分离度不足等情况,参考已有液体饮料中7种植物毒素的液相色谱测定方法[9]进行优化,固定甲醇为流动相的一种,考察水、乙酸铵缓冲液、甲酸溶液等不同流动相比例,以出峰时间、峰形及峰分离度为指标考察流动相比例。同时,液相色谱柱的填充物对混合标液分离效果也有较大的影响,对比了Wlech Xtimate C8和Waters WAT054275 C18色谱柱的效果,对液相色谱的参数、室内温度等进行调整,让液相色谱仪处于最佳状态,从而得到了较好的分离效果。
1.5 样品提取净化QuEChERS方法设计
1.5.1 样品提取 参考QuEChERS法用于凉茶中[19]的前处理技术,通过对比振荡提取、超声提取的加标回收率,选择合适的提取方式。
在提取阶段QuEChERS方法通常加入过饱和的无水硫酸镁,加快有机相与水相分层,且提高样品回收率。因此试验先考察加入单一除水剂无水硫酸镁的回收率,确定其最佳加入量,再比较在确定加入无水硫酸镁的基础上加入氯化钠和醋酸钠的加标回收率,确定用一种除水剂或多种除水剂。通过固定一种盐的量,变化另一种盐的量,根据回收率结果,对除水剂的用量比例进行优化,从而得到较好的除水剂比例。
1.5.2 样品净化 QuEChERS方法中,PSA、C18和GCB是常见的3种吸附剂。PSA能够清除样品共萃取物中的脂肪酸、某些色素和糖类等极性基质成分,而对生物碱无吸附作用[12]。因此试验先考察仅加入PSA 吸附剂的净化效果,再在确定加入PSA的基础上,考察加入C18和GCB吸附剂时的加标回收率,且对比使用不同质量的PSA,从而得到理想的净化条件。
1.6 数据统计与分析
摸索不同浓度的混合标准溶液在液相色谱中得到的响应值,将横轴变量设定为植物毒素浓度,并将峰面积设定为纵轴变量,从而得到标准曲线及其线性关系。基于标准曲线,通过外标方法计算加标样品中各种植物毒素的回收率,并获得购买样品中的植物毒素含量。通过阴性样品的加标回收率试验得到不同加标浓度下的回收率和精密度,采用控制变量法筛选出较优试验条件。
为验证方法的可靠性,采用加标回收法,在阴性植物固体饮料样品中,分别添加高、中、低3个质量浓度水平的10种植物毒素混合标准溶液,测量每个浓度水平6次以获得回收率和相对标准偏差。连续测试相应浓度的加标溶液5 d,并测量日间的精度。
2 结果与讨论
2.1 色谱条件的优化
2.1.1 流动相条件的优化 通过对比不同流动相及各种pH的流动相,发现对于这类碱性较强、极性较大的植物毒素在酸性较大的乙酸铵缓冲盐流动相中能有较好的分离度,当流动相pH值为3.5时能得到较好的峰状态,并当pH值低于3.5时,峰形及峰分离度等变化不大,考虑到液相色谱柱的耐酸性,最终确定以pH为3.5的乙酸铵缓冲液和甲醇作为流动相进行梯度洗脱。
2.1.2 液相色谱柱的选择 对比了Wlech Xtimate C8和Waters WAT054275 C18色谱柱,发现色谱柱Wlech Xtimate C8(4.6 mm×250 mm,5 μm)有更好的分离度。
2.1.3 最佳液相色谱条件的确定 将10种植物毒素混合标准溶液用Wlech Xtimate C8色谱柱,以pH为3.5的乙酸铵缓冲液和甲醇作为流动相进行液相色谱检测,调节流动相比例,通过液相色谱DAD检测器全波段扫描,得到了全波段扫描图,通过筛选分别在260,290,355 nm处得到了较好的标准品色谱图,如图1所示。
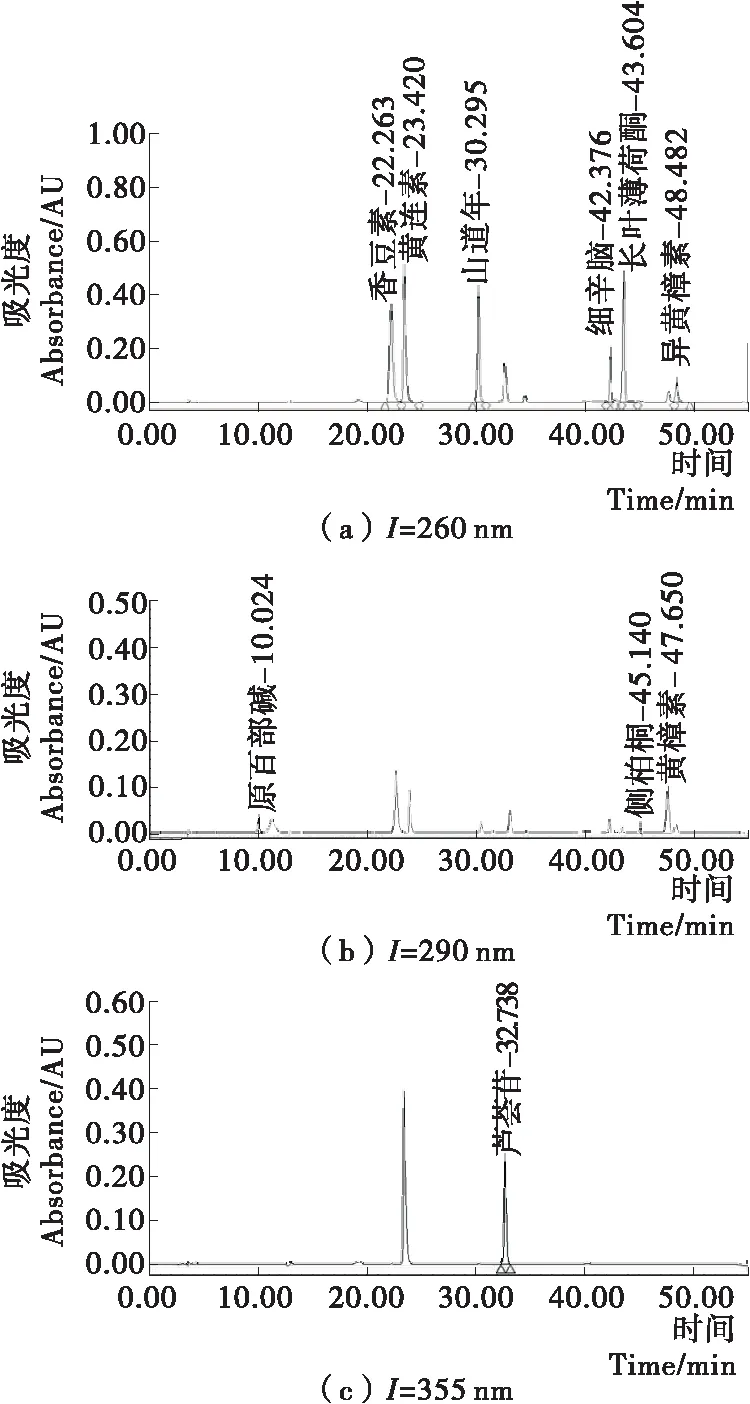
图1 10种植物毒素化合物的高效液相色谱图
最终确定最佳的色谱条件:色谱柱为Wlech Xtimate C8(4.6 mm×250 mm,5 μm);流动相A为10 mmol/L乙酸铵溶液(pH 3.5),流动相B为甲醇;梯度洗脱程序:0~15 min,70% A;15~20 min,70%~60% A;20~35 min,60%~45% A;35~36 min,45%~35% A;36~50 min,35% A;50~51 min,35%~70% A;51~55 min,70% A。流速0.8 mL/min;柱温35 ℃;后运行时间5 min;检测波长260,290,355 nm;进样量10 μL。
2.2 样品处理条件的优化
2.2.1 提取条件 通过对比振荡提取、超声提取,发现超声提取操作简单,且比振荡提取有更高的提取率。乙腈极性较强,与水互溶,加入盐后容易与水分离,与丙酮和乙酸乙酯相比,乙腈不易提取色素和基质中的鞣质、蛋白质等成分,因此选择乙腈作为提取液。
当仅用无水硫酸镁作除水剂时,加入约50 μg/L混标,随着无水硫酸镁用量的增加,能较好地除去样品中的水分,其加标回收率也逐渐增大。如图2所示,当无水硫酸镁用量>2 g时,过多的无水硫酸镁吸收水分后会吸收其他有效成分,导致回收率有所降低,因此无水硫酸镁用量为2 g时较理想。
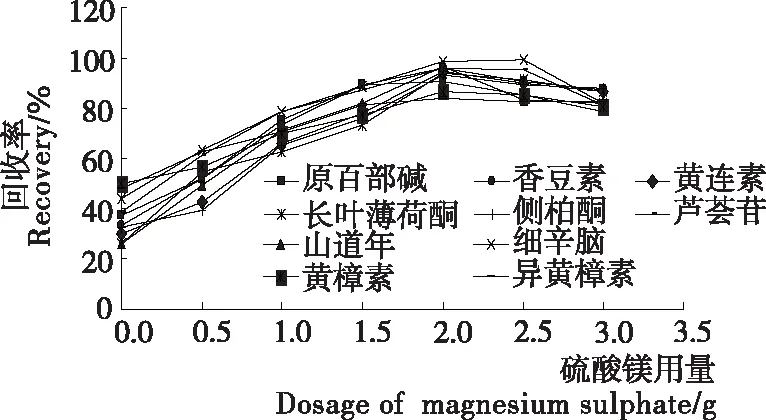
图2 无水硫酸镁用量对10种植物毒素回收率的影响
在确定加入无水硫酸镁的基础上比较加入氯化钠和醋酸钠的效果,发现加入氯化钠的回收率较高。因而氯化钠在该方法中吸水效果较好,通过试验,固定无水硫酸镁的量为2 g,变化氯化钠的量,对无水硫酸镁和氯化钠的用量比例进行优化,结果(图3)表明:当氯化钠的质量为2 g时,回收率水平最为理想,表明在无水硫酸镁与氯化钠以1∶1混合时,前处理所加水分被较好除去且不损失其他成分。

图3 除水剂组成比例对10种植物毒素回收率的影响
2.2.2 净化条件 对比3种吸附剂PSA、C18和GCB的吸附效果,发现用多种吸附剂的回收率均不如仅用PSA的,随着PSA中加入C18和GCB比例的增加混标回收率呈下降趋势(见图4),印证了PSA不仅能够清除脂肪酸、色素和糖类等成分,对生物碱无吸附作用,有较高的保留该混合溶液中植物毒素的效果。对PSA的添加量进行对比,得知用400 mg PSA作为吸附剂回收率最高,见图5。在QuEChERS方法中虽然较多的吸附剂能有较好的吸附效果,但并不是吸附剂添加越多越好,随着吸附剂的增加上清液越来越澄清,但吸附剂添加至一定量时会出现饱和状态,回收率不再随着吸附剂的增加而增加,甚至回收率会有所降低。
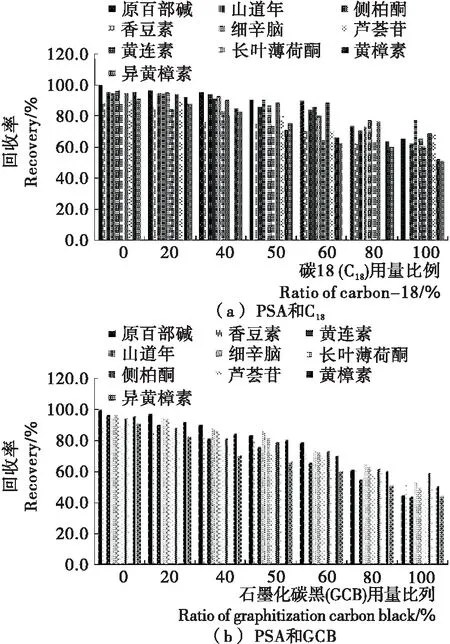
图4 吸附剂比例对10种植物毒素回收率的影响

图5 吸附剂PSA含量对10种植物毒素回收率的影响
最终确定样品前处理方法:称取5.000 g固体饮料置于50 mL具塞离心管中,加10 mL水和15 mL乙腈超声15 min。加入2 g无水MgSO4,2 g NaCl,涡旋混匀2 min,于8 000 r /min离心5 min。准确吸取上清液5 mL置于已加入400 mg PSA的50 mL离心管中,混匀,8 000 r/min离心10 min。准确吸取上清液过0.45 μm有机滤膜于样品瓶中,供液相色谱分析测定。
2.3 标准曲线、线性关系与检出限定量限
由表1可知,10种植物毒素在固定的浓度范围内其相关系数均大于0.997,表示其均有较好的线性关系,适用于固体饮料中10种植物毒素的定量检测,由于标准品均为固体配置而成,因而线性范围并非整数。在阴性固体饮料样品中添加不同浓度的待测物,按照试验建立的方法进行测定,得出10种植物毒素的检出限(S/N=3)为0.010~0.270 mg/kg,定量限(S/N=10)为0.032~0.830 mg/kg,说明该方法检测浓度较低,具有较好的灵敏度,能够满足日常检测检验。
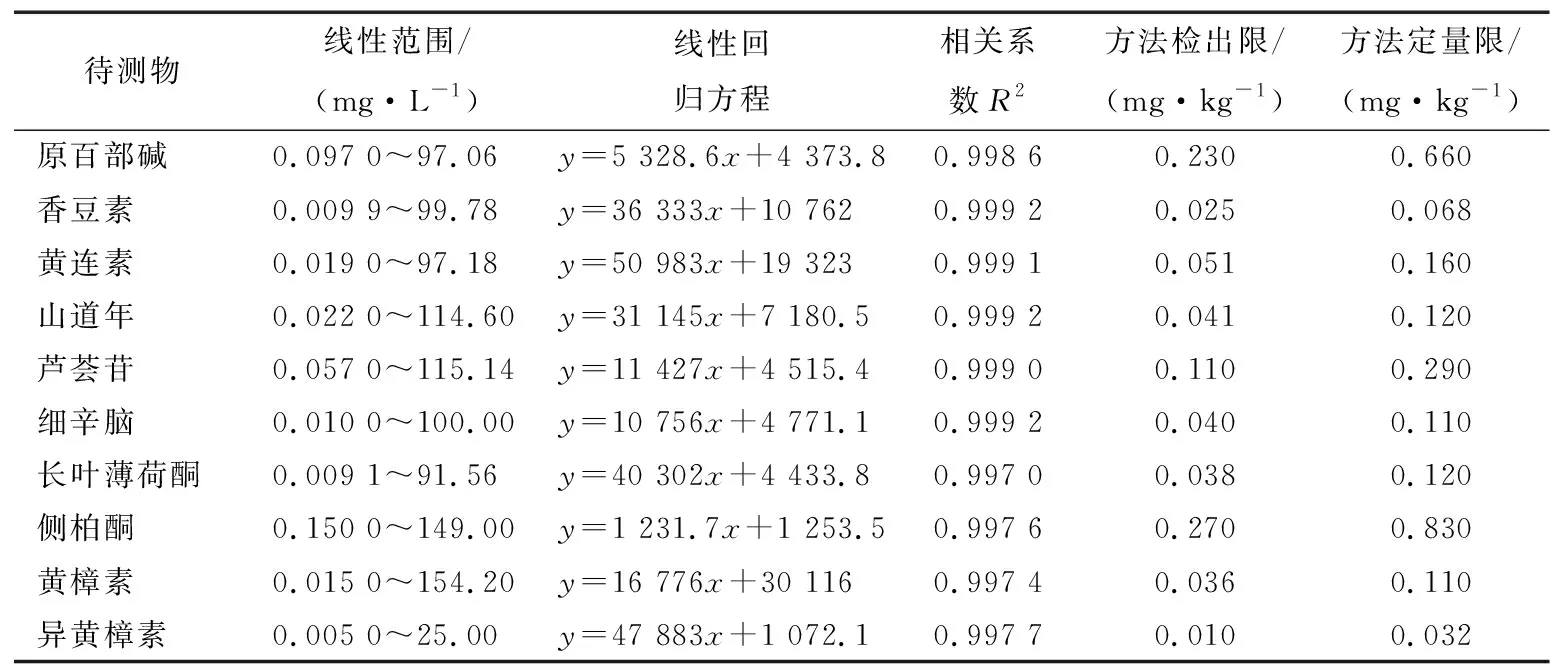
表1 10种植物毒素化合物的线性方程、相关系数、检出限和定量限
2.4 方法的准确度和精密度
由表2可知,加标回收率最低77.2%,最高105.2%,且RSD值为1.02%~2.96%,浓度较低的回收率较低,浓度高的回收率较高,添加较低浓度水平的加标样在处理过程中损失的可能性更大。这些加标样回收率的RSD值变化幅度并不大,表示该方法稳定性好,侧柏酮的响应值是所有待测物中最低的,在检测的过程中会有少许波动,因此其回收率RSD值变化稍大一些。

表2 回收率与精密度的结果
试验范围内10种植物毒素化合物的日间精密度为1.21%~2.85%,且波动范围均不大,表明该仪器稳定性好,精密度高。得到的数据均能达到GB/T 27404—2008《实验室质量控制规范食品理化检测》的要求,能够满足日常检测需要。
2.5 实际样品的测定
用试验建立的前处理方法处理10种市售植物固体饮料,并通过建立的液相色谱方法对所有样品进行DAD扫描,发现其中有一个混合类代餐固体饮料中含有香豆素,其浓度为0.04 μg/L,结合样品处理过程,得到其含量为0.20 mg/kg,尽管该结果低于《欧盟食品中化学污染物限量规定》对于植物性的饮料里植物毒素香豆素≤2 mg/kg的限量规定,但表明目前市售的植物固体饮料存在潜在的风险。
3 结论
将QuEChERS样品处理方法用于植物固体饮料中植物毒素原百部碱、侧柏酮、香豆素、黄连素、黄樟素、山道年、芦荟苷、细辛脑、异黄樟素、长叶薄荷酮的测定,通过对QuEChERS处理方法以及液相色谱条件的优化,建立了定性、定量检测植物固体饮料中10种植物毒素的检测方法。选择乙腈提取样品中的植物毒素,应用QuEChERS技术净化,采用配备PDA检测器的高效液相色谱仪进行分离测定,外标法定量。该处理方法简单,纯化效果好,检测灵敏度高,对固体饮料净化效果较好,可用于植物固体饮料中10种植物毒素的定性和定量分析。样品处理结果表明,除微量香豆素外,其他植物毒素并未检出。表明植物毒素对食用者身体健康影响的潜在风险是存在的,因此建议将植物毒素的检测纳入固体饮料质量控制范畴,促进其不断规范和提高,从而提高固体饮料整体质量水平。
