贵金属在Ag2S纳米颗粒中由内向外的迁移现象
2020-07-23刘丹叶陈东刘卉杨军
刘丹叶 ,陈东 ,刘卉 ,*,杨军 ,*
1中国科学院过程工程研究所,多相复杂系统国家重点实验室,北京 100190
2中国科学院大学,北京 100049
1 引言
当材料的几何尺寸进入到纳米尺度时,就会呈现出明显区别于块体材料的物理和化学性质,这些特性在光学1-3、催化4-8、影像9-11和医药12-15等领域的应用已经受到人们的广泛关注和研究。迄今为止,纳米尺度材料尤其是异质结构纳米颗粒仍然是热点研究领域,具有巨大的活力和吸引力,大量的新现象和新规律还有待发现,充满了原始创新的机会。以金(Au)和硫化银(Ag2S)纳米颗粒室温下的融合为例,一般来说,颗粒间的融合需要一些外因诱发,如淬火、干燥或超声处理,且融合过程多发生在由相同化学组分构成的颗粒分散体系中16,17,化学组分迥异的纳米颗粒间融合的事例则非常鲜见。Qu等发现在Au与Ag2S构成的物理混合胶体溶液中,Au与Ag2S纳米颗粒能够在室温下发生融合,导致Ag2S-Au异质二聚体的形成18。他们用透射电镜(TEM)追踪颗粒物理混合后的不同阶段,给出了融合过程的机制:颗粒在胶体溶液中进行布朗运动时可能导致充分靠近,这时电子从Au向Ag2S发生转移,Au原子变为离子释放进溶液,随后又从Ag2S颗粒表面捕获电子,变回Au原子并附着在Ag2S颗粒表面。这一过程重复进行,最终导致混合胶体中独立的Au和Ag2S纳米颗粒消失,取而代之的是形成Ag2S-Au异质二聚体。
本文描述并讨论一种同样发生在纳米尺度材料中新奇现象,即贵金属在Ag2S纳米颗粒中由内向外的迁移或称扩散现象,迁移的最终结果使开始时贵金属处于内核而Ag2S处于外壳层的核壳结构纳米颗粒演变成由Ag2S和贵金属构成的异质纳米二聚体结构,和Au与Ag2S室温下融合后的结果相似。这种迁移现象和一些文献报道的Au向半导体纳米颗粒内部扩散形成鲜明对比。例如,Banin等19和Manna等20分别报道了Au从砷化铟(InAs)和碲化铅(PbTe)纳米颗粒表面向颗粒内部的扩散现象,扩散后形成Au-InAs和Au-PbTe核壳结构纳米颗粒,但原来晶型很好的半导体InAs和PbTe由于Au的扩散呈现无定型状态。可以预见,本文描述的科学现象及其所依托的机理不仅能够满足科研人员的兴趣和与生俱来的好奇心,而且在挖掘纳米尺度材料的深度应用及开发纳米材料新的合成途径方面也具有一定的意义。
2 实验部分
2.1 实验试剂
本文用到的化学试剂如氯化金(AuCl3,99%)、硝酸银(AgNO3,99%)、乙酰丙酮铂(Pt(acac)2,99%)、氯化钯(PdCl2,99%)、元素硫(S,99.5%)、油胺(80%-90%)、甲苯(99.5%)和甲醇(99%)均购自北京化学试剂公司,使用中未经进一步纯化。
2.2 样品制备
核壳结构Au@Ag2S和Pt@Ag2S的制备基于种子生长法先制备Au@Ag和Pt@Ag核壳结构纳米颗粒,然后通过和元素硫反应将Ag壳层转变成Ag2S壳层。详细地,将20 mL油胺置于一个体积为100 mL的三口烧瓶中,向其中加入30.3 mg氯化金或混有3.5 mg硝酸银的39.3 mg乙酰丙酮铂,在氮气保护和搅拌状态下用油浴加热的方式升温至160 °C并保持1 h,以便Au3+离子或Pt2+离子能够被油胺还原成为Au或Pt原子并聚集形成Au或Pt纳米颗粒(混入少量AgNO3是为了得到近球形的Pt纳米颗粒,单纯的Pt(acac)2在油胺中还原将得到枝状结构的Pt颗粒)。之后,快速加入17 mg硝酸银,继续在氮气保护和搅拌状态下保持160 °C、3 h以完成Ag壳层在Au或Pt种子颗粒表面的生长。
反应后,将反应体系的温度降至80 °C,然后向其中加入15 mg的元素硫,维持80 °C的反应温度、3 h,将Au或Pt种子颗粒表面的Ag壳层转化为Ag2S壳层。之后,将体系降至室温,向其中加入50 mL甲醇沉淀形成的Au@Ag2S或Pt@Ag2S核壳结构纳米颗粒,离心收集,再用甲醇洗涤两次,最后重新分散于20 mL甲苯中。
核壳结构Pd@Ag2S纳米颗粒的制备采用一种不同的方式,先制取Ag纳米颗粒作为模板,然后与氯化钯进行伽伐尼置换得到Ag/Pd二元合金纳米颗粒,进而与元素硫反应将合金中的Ag转换成为Ag2S并包覆在Pd的表面21。具体的,将17 mg硝酸银加入到20 mL油胺中,160 °C下反应1 h得到分散于油胺中Ag纳米颗粒,然后向其中加入17.7 mg的氯化钯,保持温度为160 °C继续反应2 h;反应后体系温度降至室温,用甲醇沉淀形成的Ag/Pd合金颗粒,离心收集并洗涤两次后分散于20 mL甲苯中;进而在分散有Ag/Pd纳米颗粒的甲苯中加入15 mg元素硫,搅拌状态下放置5 h以便合金中的Ag能被充分萃取、反应并在Pd表面形成Ag2S壳层。最后,重复甲醇沉淀、离心收集和洗涤的过程,获得的Pd@Ag2S纳米颗粒再重新分散于20 mL甲苯中。
核壳结构Ag@Ag2S纳米颗粒的制备比较简单,同样是先在油胺中制备Ag纳米颗粒,反应条件和前面相同。同时,将3.2 mg元素硫溶解在10 mL油胺中,然后取250 μL加入预先制好的Ag胶体溶液中,并在160 °C下保持搅拌10 min。因加入的元素硫不足以全部将Ag转换成Ag2S,它只是将位于表层一定深度的Ag原子转化成Ag2S,并包覆在未反应的Ag内核表面,形成Ag@Ag2S核壳结构纳米颗粒。反应后,经甲醇沉淀、离心收集,两次甲醇洗涤,最后重新分散在20 mL甲苯中。
这些最终分散在甲苯中的Au@Ag2S、Pt@Ag2S、Pd@Ag2S以及Ag@Ag2S核壳结构纳米颗粒在室温下静置72 h,并间或用透射电镜追踪颗粒的结构演变过程,分析贵金属在Ag2S中的迁移情况。
2.3 样品表征
核壳结构纳米颗粒的形貌、尺寸和结构演化过程采用工作电压为200 kV的场发射透射电子显微镜(transmission electron microscope,TEM,JEOL JEM 2100F,Tokyo,Japan)进行表征。样品制备过程是将一片直径为3 mm且覆盖有连续碳膜的铜网正面向上放置于一张滤纸表面,向铜网滴加一滴样品胶体溶液,多余的溶剂被滤纸自然吸收,然后在室温下进行真空干燥。贵金属迁移前后样品胶体溶液的紫外光谱用Hitachi U-3900分光光度计(Hitachi,Japan)获取。颗粒样品的晶相结构用Bruker D8 X射线衍射仪(X-ray diffraction,XRD,Bruker AXS,German)进行表征。样品制备过程为首先用甲醇沉淀颗粒,离心收集并用甲醇多次洗涤后真空干燥。
3 结果与讨论
图1a为基于种子生长法制备的Au@Ag核壳结构纳米颗粒,颗粒的直径约为15.4 nm。由于Au和Ag元素的原子量差异较大,透射电镜(TEM)下内核和壳层有较为明显的衬度差别,Au处于内核位置而Ag处于壳层位置,如图1a右下角插图所示意。图1a右上角插图是一个Au@Ag单颗粒的高分辨透射电镜(high-resolution TEM,HRTEM)图像,显示Ag壳层在Au内核颗粒表面有外延生长的趋势。

图1 核壳结构Au@Ag (a)、Au@Ag2S (b)及贵金属迁移后形成Au-Ag2S异质结构(c)和迁移中间状态时(d)的透射电镜图像。右上插图为相应的高分辨透射电镜图像;右下插图为各阶段相应的结构示意图Fig.2 TEM images of core-shell Au@Ag (a), core-shell Au@Ag2S (b), heterogeneous Au-Ag2S due to Au migration (c) and intermediate structures during Au migration (d).Insets in the top and bottom right-hand corners are corresponding HRTEM images and schemes of the nanostructures in each stage, respectively.
通过和元素硫反应,核壳结构Au@Ag颗粒表面的Ag壳层被转换为Ag2S壳层,最终导致形成Au@Ag2S核壳结构纳米颗粒,其TEM图像如图1b所示。和最初的核壳结构Au@Ag颗粒相比,转化后Au@Ag2S核壳结构纳米颗粒的直径有所增加,约为18.8 nm。由于半导体材料相比于贵金属电子密度较低22,23,Ag壳层转换为Ag2S壳层后TEM图像上和Au内核的衬度差别更加明显,核壳结构很容易辨识,其示意图如图1b右下角插图所示。一个核壳结构Au@Ag2S纳米颗粒的HRTEM图像如图1b的右上插图所示,可以观察到,Ag转变为Ag2S后,壳层沿内核Au颗粒晶格外延生长的趋势变得不明显。
为了证实核壳结构中的Ag壳层已经转化成为Ag2S壳层,我们分析了和元素硫反应前后颗粒的X射线衍射(XRD)图谱。如图2a所示,由于Au和Ag都有相同的面心立方(FCC)晶体结构,且结构参数相近,核壳结构Au@Ag的XRD图谱只有一套衍射峰,从左至右5个峰分别对应晶体的(111)、(200)、(220)、(311)和(222)晶面;Ag壳层和元素硫反应后,最终产物的XRD图谱变化较大,除了保留了Au内核的衍射峰外,其余的衍射峰则可归属于具有单斜晶体结构的Ag2S相,证实核壳结构Au@Ag中的Ag壳层成功向Ag2S转变。这一转变也可以通过和元素硫反应前后Au@Ag胶体溶液的紫外光谱进行说明。如图2b所示,和元素硫反应前,核壳结构Au@Ag胶体溶液有一个很宽的紫外吸收峰,中心在波长450 nm处,而和元素硫反应后,由于Ag2S不具有紫外吸收,生成的核壳结构Au@Ag2S纳米颗粒只显示Au内核的紫外吸收特性,其吸收峰中心红移到波长为530 nm处。
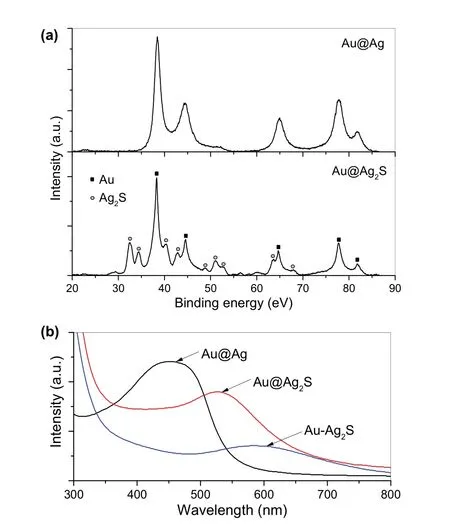
图2 核壳结构Au@Ag和Au@Ag2S的XRD图谱(a),核壳结构Au@Ag及Au迁移前后的Au@Ag2S的紫外吸收光谱(b)Fig.2 XRD patterns of core-shell Au@Ag and Au@Ag2S nanoparticles (a), UV-visible spectra of core-shell Au@Ag as well as Au@Ag2S before and after Au migration (b).
实验观察发现,形成的Au@Ag2S核壳结构纳米颗粒是不稳定的,Au能够在核壳结构中由内向外发生迁移。图1c及其右上和右下插图分别是Au@Ag2S核壳结构纳米颗粒在甲苯中静置72 h后的TEM图像、HRTEM图像和结构示意图。通过和图1b进行比较,可以清晰地判定静置后Au已经由最初的内核位置迁移至Ag2S的表面,并和Ag2S构成了具有异质结构的Au-Ag2S纳米颗粒。Au迁移后形成的Au-Ag2S异质二聚体胶体溶液的紫外谱图如图2b中蓝线所示,其吸收峰中心和Au迁移前的核壳结构Au@Ag2S相比,进一步红移到600 nm,显示Ag2S在Au表面的不同包覆方式对其光学性质有巨大影响。这一现象并非首次发现,早在2010年,Yang和Ying就已经报道了纳米尺度上Au在Ag2S颗粒中由内向外的扩散现象24。他们在水相制取Au纳米颗粒,然后借助十二胺和乙醇的辅助转移入甲苯,并和预先用同样方法转移入甲苯的Ag+离子混合,然后室温下加入元素硫,在Au颗粒表面形成Ag2S壳层。他们发现经过一段时间放置,Au能够从内核位置扩散至表面,形成Au-Ag2S二聚体结构。当时的解释是由于Au和Ag2S能带结构的差异,Au内核原子的电子转移至Ag2S,然后Au离子通过空位机制穿越Ag2S扩散至外表面,然后再捕获通过间隙机制扩散出来的电子并附着在Ag2S壳层的表面。这一机制的核心是扩散在所有方向上均匀进行,扩散出来Au原子最后经过一个熟化过程在Ag2S表面形成一个大颗粒。
然而,通过对扩散中间状态的表征发现,在本文制取的核壳结构Au@Ag2S纳米颗粒中,Au在Ag2S中由内向外的迁移类似于整体进行而非先是在各个方向上均匀扩散然后再进行熟化。图1d及右上插图是核壳结构Au@Ag2S纳米颗粒在甲苯中静置36 h后的TEM和HRTEM图像,可以明显观察到Au在迁移至表面之前在Ag2S中整体向一端移动,且由于Au的整体迁移,原本呈球形的核壳结构纳米颗粒演变成了近似椭圆形,如图1d右下插图所示意。
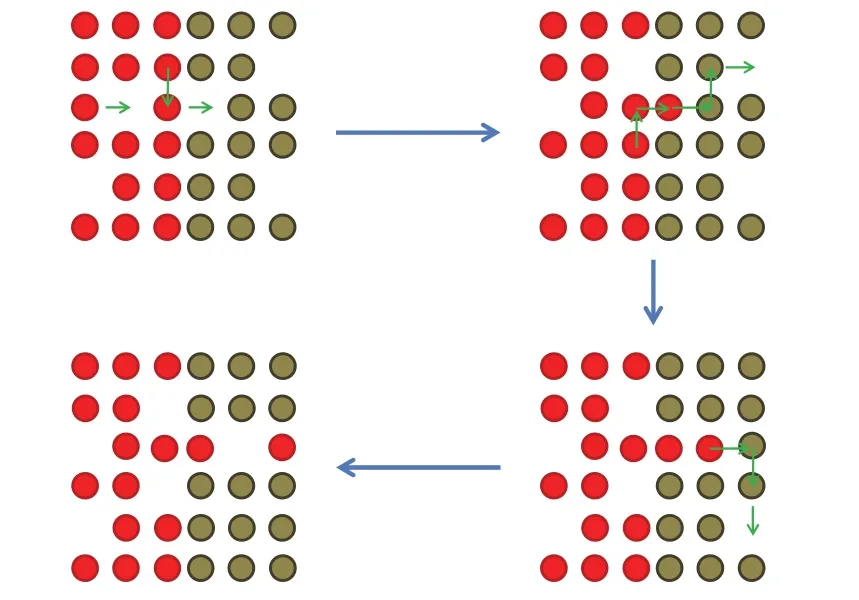
图3 贵金属在Ag2S纳米颗粒中由内向外迁移示意图Fig.3 Schematic illustration showing the inside-out migration of noble metals in Ag2S nanoparticles.
有两种机制可以解释这一现象,如图3所示:其一是将Au内核和Ag2S壳层看作一对扩散偶。核壳结构Au@Ag纳米颗粒中的Ag壳层转化为Ag2S壳层后,由于单斜相的Ag2S壳层与面心立方相的Au内核晶体结构差异较大,相界面处失配严重,界面质点(原子、离子)必然处于较高的能量状态,容易脱离各自化学键的束缚而迁移进入另一相。一定温度下,晶体中均存在平衡浓度的点缺陷,如空位等,这些点缺陷尤其空位可以协助质点迁移以换位的方式进行。但Au和Ag2S构成的扩散偶中,Ag+离子向Au中的迁移可能性较低,因为要保持局部电中性,Ag+离子的迁移必然伴随着S2-的迁移,这种状况不容易发生。因此,扩散偶中发生的迁移主要是Au原子在Ag2S中以空位机制进行的扩散。这一迁移过程一旦在核壳结构Au@Ag2S颗粒内部某一方向引发,Au内核的其它原子便会遵循这一方向Au原子迁移产生的空位继续迁移,便导致观察上Au内核在Ag2S中整体移动的效果。
其二是将Au内核视作掺杂在Ag2S半导体中的杂质,然后被半导体Ag2S纳米颗粒以一种自纯化(Self-purification)机制排出体外。异类原子掺杂能够导致实际晶体中的某些区域与理想晶体发生偏差,是一类晶体点缺陷,而半导体为保持自身质点排列的规则性,有排出掺杂在其中的异类原子的趋势,称为自纯化机制,这一机制经常被用来解释向半导体中掺杂金属或非金属原子所遇到的困难25,26。Turnbull曾经报道尺寸小的颗粒其自纯化机能更加强烈,从而导致其自身的点缺陷更少27。这是因为,随着颗粒的尺寸减小,点缺陷包括异类原子更加容易排出,它们在晶体中迁移至表面所经历的距离要远远小于块体材料。Dalpian和Chelikowsky从能量的角度以热力学的观点解释了这一现象,他们认为自纯化是半导体材料的一种固有性质并且发现缺陷形成能随着颗粒尺寸的减小快速增加,因而小颗粒有更加强烈的意愿排出掺杂在自身中的异类原子28。基于这一机制,在核壳结构Au@Ag2S纳米颗粒中,由于面心立方Au内核和单斜Ag2S壳层间的严重晶格失配,处于高能量状态的Au原子一旦摆脱金属键的束缚,通过和界面处的空位交换迁移进入Ag2S相,就可以被处于纳米尺度上的Ag2S以自纯化机制排出至表面,这一过程重复进行也可造成观察上Au内核在Ag2S中沿着某一方位整体迁移至颗粒表面。

图4 核壳结构Ag@Ag2S (a)、Pd@Ag2S (c)和Pt@Ag2S纳米颗粒(e)及贵金属迁移后形成的Ag-Ag2S (b)、Pd-Ag2S(d)和Pt-Ag2S异质结构纳米颗粒(f)的透射电镜图像Fig.4 TEM images of core-shell Ag@Ag2S (a), Pd@Ag2S(c), and Pt-Ag2S nanoparticles (e) as well as heterogeneous Ag-Ag2S (b), Pd-Ag2S (d), and Pt-Ag2S nanostructures (f)due to migration of noble metals.
这种贵金属在Ag2S纳米颗粒中的迁移现象并非Au所独有,本文通过制备核壳结构Ag@Ag2S、Pd@Ag2S和Pt@Ag2S纳米颗粒并观察对比在甲苯中静置前后颗粒的透射电镜图像,发现涉及的贵金属如Ag、Pd和Pt均能由内核位置迁移至Ag2S颗粒的外表面,并与Ag2S构成异质二聚体结构。图4a,c和e为制取的核壳和结构Ag@Ag2S、Pd@Ag2S和Pt@Ag2S纳米颗粒的TEM图像,由于尺寸较小的原因和电子密度差异的不同,它们内核和壳层的衬度差别不似Au@Ag2S那样明显,但同样可以清晰辨识。图4b,d和f是同一样品静置48 h后的TEM图像,它们和静置前核壳结构的图像有显著差别,具有明显的异质二聚体结构,分别为Ag-Ag2S、Pd-Ag2S和Pt-Ag2S。需要注意的是,表征发现Ag、Pd和Pt在Ag2S中由内向外迁移要比Au快的多,甚至难以观察到中间状态,这和核壳结构纳米颗粒的整体尺寸以及异类原子(Ag、Pd或Pt)的尺寸有关,整体尺寸越小,异类原子半径越小,迁移越容易进行。在本文研究中,核壳结构Ag@Ag2S、Pd@Ag2S和Pt@Ag2S纳米颗粒的直径分别约为11.2、9.4和5.6 nm,远小于Au@Ag2S核壳颗粒的直径(18.8 nm),而Ag、Pd和Pt原子的半径分别144、138和136 pm,和Au原子半径(144 pm)相当或略小,综合这些因素,导致Ag、Pd和Pt原子在Ag2S中发生相比Au原子较快的迁移。
贵金属在半导体中的迁移可以用来制备一些常规方法不易获得的异质结构纳米材料,比如Pd-Ag2S和Pt-Ag2S异质二聚体,用常规种子生长法在Ag2S颗粒存在时还原Pd和Pt金属前驱体通常很难成功,要么贵金属在Ag2S表面多个位点生长(水相制备)29,要么形成连续的贵金属壳层包覆整个半导体种子颗粒(有机相制备)30,而贵金属在半导体中的迁移则提供了一种可行的选择。
4 结论
本文利用透射电镜观察,结合X射线衍射和紫外光谱辅助手段,描述并讨论了贵金属如Au、Ag、Pd和Pt在Ag2S纳米颗粒中由内向外的迁移现象。迁移可在室温下进行,其最终结果使开始时贵金属处于内核而Ag2S处于外壳层的核壳结构纳米颗粒(Au@Ag2S、Ag@Ag2S、Pd@Ag2S和Pt@Ag2S)演变成由贵金属和Ag2S构成的异质纳米二聚体结构,如Au-Ag2S、Ag-Ag2S、Pd-Ag2S和Pt-Ag2S。研究发现,在实验条件下贵金属在Ag2S的迁移并非在各个方向上均匀进行,而是类似于一种整体迁移的模式,迁移过程中球形的核壳结构颗粒有向椭圆形演变。贵金属在Ag2S中的经空位互换的扩散机制或半导体纳米颗粒的自纯化机制可以用来解释这种迁移现象。本研究揭示了一种发生在纳米尺度材料中的新奇现象,可以设计制备一些常规过程难以获取的异质结构纳米颗粒,在开发纳米材料有效合成途径方面也具有一定的价值。
猜你喜欢
——庆祝中国共产党成立一百周年贵金属纪念币展
