Signaling pathways downstream to receptor tyrosine kinases: targets for cancer treatment
2020-07-21EmmaCordoverAudreyMinden
Emma Cordover, Audrey Minden
Susan Lehman Cullman Laboratory for Cancer Research, Department of Chemical Biology, Ernest Mario School of Pharmacy, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA.
Abstract Mammalian cells have the ability to respond to a myriad of diverse extracellular stimuli that modulate cell function. This often involves ligands binding to cell surface receptors and subsequent activation of intracellular signaling pathways. These pathways can lead to changes in gene expression patterns that in turn regulate cell growth, differentiation, migration, and function. One important type of cell surface receptor is the receptor tyrosine kinase (RTK). In response to in response to ligand binding, RTKs dimerize, then trans-phosphorylate each other, leading to activation of downstream pathways. While the signaling proteins in these pathways are important for normal cell growth control, when improperly regulated they can lead to uncontrolled growth and sometimes cancer. For this reason, they are often considered to be good candidates for drug targets for chemotherapeutic drugs. RTKs can activate multiple different signaling pathways. Some of the signaling proteins in these pathways can have crosstalk with other RTK activated pathways, and some of them can be activated by multiple mechanisms in addition to activation by RTKs. While there is a wide array of different signaling proteins and pathways activated by RTKs, in this review we will discuss components of several key pathways including the MAPK pathway, the Her2/Neu pathway, mTOR, and Pak kinases. We provide an overview of the roles for these pathways in cell signaling and discuss how different components of these pathways are being considered as targets for cancer treatment.
Keywords: Receptor tyrosine kinases, signaling pathways, targeted cancer therapeutics, oncogenes, cell signaling
INTRODUCTION
Cells within multicellular organisms depend upon complex ways of communicating with their surroundings and other cells and tissues. An important method by which cells communicate is by responding to signals from extracellular stimuli. Growth factors are small extracellular ligands that transmit signals between cells and their environment. They can control cell function by binding to transmembrane receptors, activating a myriad of different intracellular signaling pathways. Growth factors commonly bind to the receptor tyrosine kinase family of transmembrane receptors (RTKs). RTKs have an extracellular domain that binds to ligand, a transmembrane domain, and an intracellular domain that has tyrosine kinase activity. Once bound to ligand, the RTKs dimerize, and transphosphorylate on tyrosine residues. This usually leads to binding of adapter proteins to the phospho-tyrosine residues, leading in turn to a cascade of events within the cell[1]. This includes activation of GTP binding proteins, a cascade of serine/threonine phosphorylation events, and translocation of certain proteins to the nucleus. Ultimately this can lead to the regulation of transcription factors which in turn result in changes in gene expression patterns. The result is the regulation of genes that control cell growth, differentiation, survival, and function[2]. In order to understand how normal cell function and communication is regulated, it is critical to understand how those intracellular events occur in normal cells.
In addition to their important roles in controlling normal cell fate, it is also important to understand the roles that signaling pathways play in cancer. Intracellular signaling pathways are often improperly regulated in cancer[3]. As a result, cancer cells often become independent from their control by extracellular stimuli. Instead, they can gain the ability to grow, proliferate, and survive, without external regulation. This is an important feature of many cancer cells, and one that is important to understand in order to develop individualized treatments.
Understanding which intracellular signaling proteins are most important for controlling cellular behavior is important for understanding the molecular basis of cancer. There are multiple types of extracellular stimuli and intracellular signaling pathways that are important for controlling cell growth. These include not only pathways regulated by RTKs, but also pathways that are regulated by other types of receptors such as trimeric G protein coupled receptors and cytokine receptors. The study of RTKs has taken a prominent role in studying cancer. This is in part due to being linked to the Ras family of proteins, which are mutated in a large percentage of cancers[4]. Furthermore, multiple components of RTK activated signaling cascades have been linked to cancer when they are aberrantly regulated. In this review we will focus on the signaling pathways that are mediated by RTKs, which have been implicated in cancer when improperly regulated. While the number of diあerent pathways that are associated with cancer is large, we will focus on several RTK regulated pathways in this review, and we will discuss their importance in cancer research. We then discuss some of the drugs and drug candidates that are designed to target signaling proteins within these pathways.
MITOGEN-ACTIVATED PROTEIN KINASE PATHWAY
The mitogen-activated protein kinase (MAPK) signaling pathway was discovered over 30 years ago. It is one of the most well studied intracellular signaling pathways and it often serves as a model for RTK activated signaling pathways. The MAPK pathway is frequently implicated in cancer and serves as an important target for cancer therapies. The MAPK signaling cascade plays several key physiological roles in healthy cells. Specifically, MAPKs mediate cellular growth, proliferation, and survival processes[5]. MAPK operates by modulating transcription factors, which in turn leads to regulation of gene expression in response to extracellular signals[6]. Abnormal MAPK signaling is shown to be associated with several types of cancer such as breast cancer, prostate cancer, colorectal cancers, melanoma, and leukemia. Dysregulation of MAPK signaling in cancer is associated with evasion of apoptosis, uncontrolled cell proliferation, and resistance to chemotherapy and targeted therapies[7].
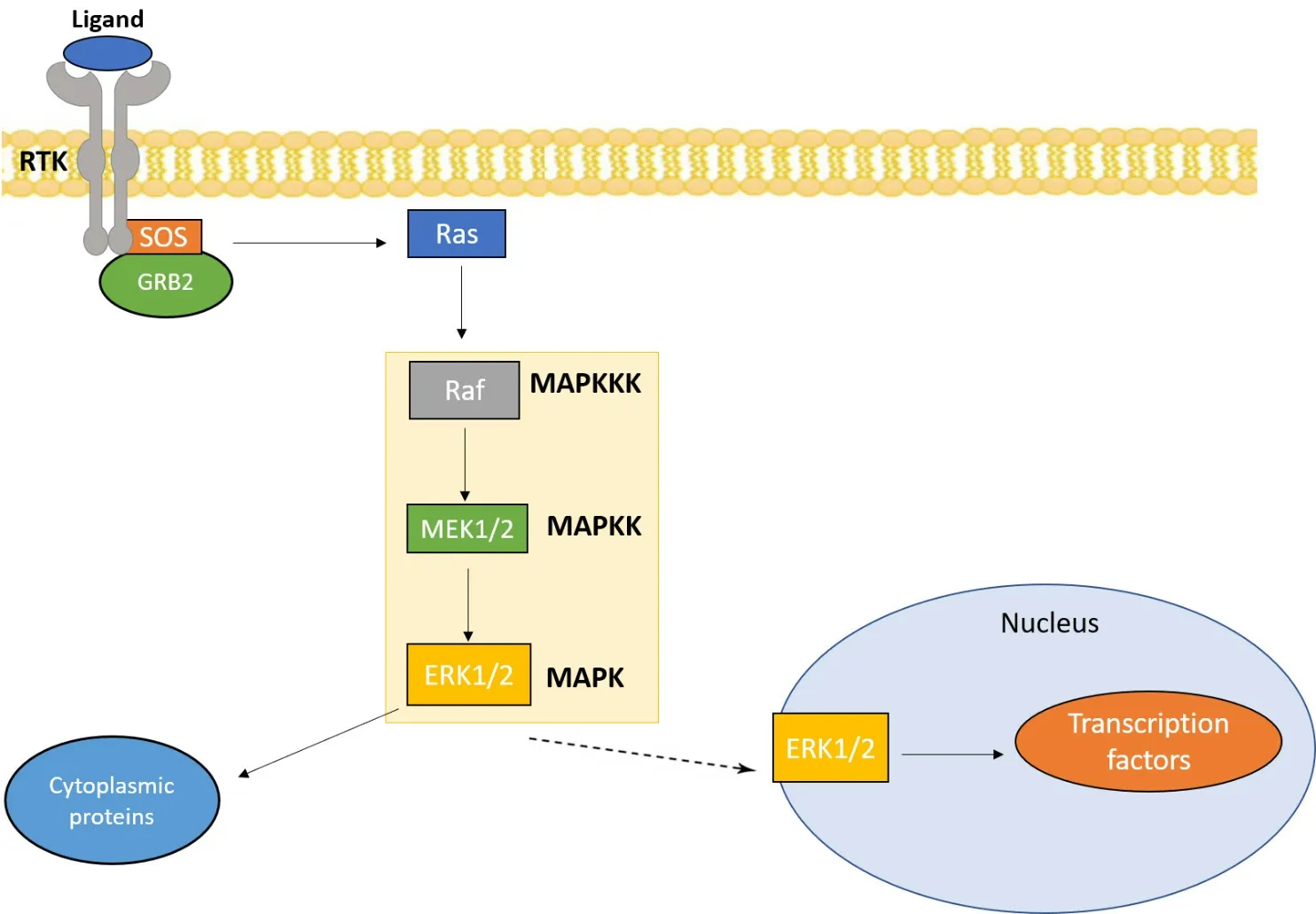
Figure 1. MAPK/ERK pathway: the MAPK/ERK pathway is activated by a variety of extracellular signals, such as growth factors and other mitogens. The main components of this pathway are Ras, Raf, and MEK which lead to the activation of ERK. ERK subsequently activates several cytoplasmic proteins and transcription factors that influence cell proliferation and survival. MAPK: mitogen-activated protein kinase; ERK: extracellular signal-regulated kinases; MAPKK: MAPK kinase; MAPKKK: MAPK kinase kinase
The MAPK pathway can be activated by multiple mechanisms, including activation of RTKs in response to extracellular stimuli. For example, binding of the epidermal growth factor receptor (EGFR), an RTK, to its ligand epidermal growth factor leads to EGFR dimerization and activation. This in turn can trigger activation of the MAPK pathway [Figure 1]. The canonical MAPK cascade is composed of three successive serine/threonine kinases: MAPK kinase kinase (MAPKKK), which phosphorylates MAPK kinase (MAPKK) which in turn phosphorylates and activates MAPK[8]. MAPKKK itself is regulated in response to small GTPases, typically Ras, which are activated by exchange factors in response to the activated RTK. In humans, the MAPK family includes the extracellular signal-regulated kinases (ERK1/2), Jun N-terminal kinases (JNK1, JNK2, and JNK3), and p38[9].
Dysregulation of the ERK pathway is found in approximately one-third of all human cancers[10]. ERK, as a MAPK, operates downstream to the Ras small GTPase. The Ras/ERK pathway is activated by various stimuli, including growth factors and mitogens. Growth factors and mitogens depend on this cascade to transmit signals for regulating gene expression which in turn leads to regulation of cell growth and apoptosis[11]. Other important components of this pathway include Raf and MEK [Figure 1]. One important way the pathway is activated in quiescent cells is through the binding of a growth factor (GF) to receptor tyrosine kinases (RTKs) in the cell membrane. The binding of GFs causes the dimerization and autophosphorylation of RTKs, which leads to the recruitment of signaling molecules including SOS, the Ras GTPase exchange factor. SOS exchanges GDP for GTP to activate Ras which then recruits Raf. Raf in turn activates MEK, culminating in activation of ERK. Activated ERK phosphorylates several substrates such as transcription factors and cytoplasmic signaling proteins which have key roles in the regulation of cell growth and function[8]. Due to the alteration of the Ras/ERK pathway in many cancers, components of the pathway are considered to be promising drug targets for cancer therapies.
Ras inhibitors
The RAS genes are among the most common oncogenes in human cancer and are of particular interest for targeting the ERK signaling cascade in cancer. The Ras family of proteins consists of three different isoforms - K-Ras, N-Ras, and H-Ras. Point mutations in all three of the RAS genes are associated with different types of cancer. For instance, K-RAS mutations are seen in over half of pancreatic tumors and N-RAS mutations are present in almost 20% of skin cancers[12]. As Ras is inactive in quiescent cells, its constitutive activation drives tumor formation. Previous efforts to target Ras in cancer therapies have proven to be unsuccessful. However, the development of new direct Ras inhibitors is a promising area of research.
One way to inhibit Ras is by targeting Ras membrane localization. In order to exert its oncogenic effects, Ras must localize to the plasma membrane. The farnesyltransferase inhibitors (FTIs) were created to block Ras membrane localization. The C-terminal CaaX box is a motif that is conserved in all Ras proteins and is essential for the addition of a farnesyl lipid group. The addition of a farnesyl group to a targeted cysteine in the CaaX motif is catalyzed by farnesyltransferase (FTase) and allows Ras to interact with the membrane[13]. FTIs have been shown to be ineffective in the treatment of solid tumors with K-RAS and N-RAS mutations, but are promising in treating certain malignancies with H-RAS mutations, such as thyroid and bladder cancers[14]. The failure of FTIs in blocking K-Ras membrane localization is attributed to K-Ras having a higher affinity for FTase than H-Ras. Additionally, K-Ras can interact with geranylgeranyltransferase (GGTase) which allows for the covalent attachment of a geranylgeranyl lipid group to the CaaX domain, thus allowing K-Ras to localize to the membrane without farnesylation by FTase. Nonetheless, researchers have designed a neo-substrate that binds to the farnesylated cysteine in the CaaX domain to prevent geranylgeranylation by GTTase and subsequently, K-Ras membrane localization[13].
Another advancement is to exploit the G12C mutation in K-Ras to suppress oncogenic activity. Targeting tumors with the K-RasG12Cmutant is of clinical importance because this mutation is frequently expressed in lung and colon adenocarcinomas[15]. In addition, there were few known mechanisms for inhibiting K-Ras activity in cancer cells until the recent discovery of a druggable pocket in K-RasG12C. This target, called the switch-II pocket, has initiated the innovation of several irreversible inhibitors to lock K-RasG12Cin its inactive GDP state. There are currently several K-RASG12Cinhibitors that are in Phase I/II of clinical trials. For instance, AMG510, developed by Amgen, interacts with the His95 small groove within the switch-II pocket. Preclinical studies have shown regression of K-RASG12Ctumors and elevated T cell infiltration at tumor sites in mice treated with AMG510[16]. Likewise, MRTX849 (Mirati Therapeutics) also binds K-Ras in the switch II pocket. MRTX849 has proved to be as effective as a K-RasG12Cinhibitor by inhibiting GTPloading[16]. AMG10 and MRTX849 have both been shown to lead to inhibition of ERK phosphorylation and signaling, thus contributing to anti-tumor activity in humans. Further work is being done to determine the efficacy of K-RasG12Cinhibitors in combination with other anticancer drugs[17].
Another area of interest is to investigate Ras dimerization as a potential therapeutic target for cancer. Previous studies show that monomeric Ras can bind Raf, but numerous studies indicate that dimerization of Ras is necessary for the activation of Raf. Under normal cellular conditions, Ras-GTP is recruited to the plasma membrane where it activates the Raf kinase domain. Raf in turn activates the MAP kinase pathway. As such, all three isoforms of Ras activate Raf, and K-Ras is the most powerful activating agent of Raf[18]. Multiple studies have found evidence for the role of Ras homodimerization in Raf activation. For instance, by using a K-Ras mutant that impairs dimerization, K-Ras dimerization was shown to be critical for the abnormal Ras signaling that is seen in many cancers. While more research needs to be done to determine the mechanism behind Ras dimerization, current data suggests that disruption of Ras dimerization has the capacity to suppress Ras-Raf signaling in cancer[19].
Raf and MEK inhibitors
The Raf family of protein kinases are central components of the ERK cascade as they interact with Ras and initiate the activation of the kinase cascade. When Raf is dysregulated, it can exert oncogenic effects, including metastasis, invasion, and uncontrolled cellular proliferation[20]. Researchers have discovered mutations that affect the catalytic activity of Raf to be present in several human tumors. Of the three different isoforms of Raf, A-, B-, and C-Raf, B-Raf was found to be commonly mutated in numerous cancers, specifically melanoma. In fact, B-Raf mutations are found in 66% of all melanomas[21]. In wild type cells, Ras prompts the dimerization of B-Raf and its subsequent activation. In cancer cells with mutated B-RAF, activating mutations cause B-RAF to be expressed constitutively, which promotes cell growth and inhibits apoptosis[22].
In particular, the B-RafV600Emutation, present in melanoma and colon cancer, is shown to drive cancer development, and serves as an important diagnostic and prognostic biomarker[23]. Inhibitors that target B-RAFV600Ehave been clinically proven to prolong the survival of advanced stage melanoma patients. Nonetheless, these agents display adverse effects, and patients often relapse due to acquired resistance. A major problem with first generation B-RAF inhibitors is that drug treatment provokes increased wildtype Raf membrane localization and B-/C-Raf dimerization. This means that when the inhibitor is under non-saturating conditions, drug bound B-Raf can dimerize with drug-free Raf, resulting in the activation of MEK and ERK in the presence of active Ras. This contributes to the limited activity of these firstgeneration inhibitors in cancers with upregulated Ras despite having the V600E B-Raf mutation. As such, this poses a challenge for next generation B-RAFV600Einhibitors. One way in which researchers are looking to combat the dilemma of Raf dimerization and drug resistance is by creating inhibitors that target both monomeric and dimeric Raf with equal affinity. These agents, known as pan-Raf inhibitors, have been shown to inhibit Raf signaling and paradoxical ERK activation in colorectal cancer and melanomas. Another approach that is being studied to combat paradoxical ERK activation is through an inhibitor that binds to the ATP binding pocket of B-Raf and impedes Raf dimerization[20].
In addition to Ras and Raf, MEK is another kinase that has an important role in the ERK pathway and serves as a promising drug target. Once it is activated, MEK phosphorylates ERK. ERK in turn plays critical roles in the regulation of cellular proliferation and apoptosis. As such, the inhibition of both isoforms of MEK, MEK1 and MEK2, have the potential to cause growth inhibition in tumors where the ERK pathway is activated. Interestingly, trametinib, the FDA approved MEK1 and MEK2 inhibitor, is shown to inhibit tumor proliferation in advanced stage melanomas with the B-RAFV600Emutation. Compared to standard chemotherapy, trametinib given as a single agent showed statistically significant improvement in terms of disease progression-free survival during treatment[24]. The other FDA-approved MEK inhibitor, cobimetinib, proved successful in blocking ERK/MAPK signaling in B-RAF and K-RAS mutated cell lines. Clinically, cobimetinib is well tolerated in patients with advanced solid tumors with the B-RAFV600Emutation[25]. Further, several preclinical and clinical studies show that the use of a MEK inhibitor alongside a BRAF inhibitor is a promising form of treatment for advanced stage melanomas. As previously mentioned, melanoma patients treated with first generation BRAF inhibitors often relapsed due to acquired drug resistance. The dual inhibition of both BRAF and MEK leads to more significant tumor response than BRAF therapy alone[26].
ERK inhibitors
ERKs are important effectors in the Ras/ERK pathway. There are two structurally similar isoforms of ERK, ERK1 and ERK2, and they belong to the MAPK family of protein kinases that are central to signal transduction. ERK is involved in the regulation of many cellular processes, such as proliferation and differentiation, and its increased activity is often implicated in several human cancers[27]. ERK is activated by its upstream effector MEK through phosphorylation [Figure 1]. ERK has roles in both the cytoplasm and the nucleus. In the cytoplasm, ERK modulates key cytoskeletal proteins that affect motility, metabolism, and cell adhesion[28]. ERK exerts its nuclear functions by dimerizing and localizing to the nucleus once it is activated. In the nucleus, ERK phosphorylates and subsequently activates a number of transcription factors that are involved in cell proliferation and survival[29]. Due to its aberrant expression in several cancers, it is being studied as a potential therapeutic option in cancers with B-Raf and N/K-Ras mutations[27].
As discussed earlier in this review, BRAF mutations are common in an array of human cancers, most notably in melanoma. BRAF inhibitors are shown to be promising in the short-term, but ultimately, are ineffective at blocking cancer cell survival and proliferation. BRAF/MEK dual inhibition is associated with an increased anti-tumor response. Unfortunately, up to 70% of tumors treated with a BRAF/MEK dual inhibition strategy experience ERK/MAPK pathway reactivation and evade treatment[30]. Hence, novel strategies that target the Ras/ERK pathway in cancers with BRAF mutations are sought. Once such novel inhibitor that targets this pathway is the ERK inhibitor ulixertinib. Recent studies report on the efficacy of ulixertinib in solid malignancies in a phase I clinical trial. Ulixertinib is a highly potent, reversible, and selective ATP-competitive inhibitor of ERK1/2. Previous evidence shows that ulixertinib reduces tumorigenesis in BRAF- and Ras-mutant xenograft models[27]. The results of the phase I clinical trial provide evidence that ulixertinib is effective in cancers with a diverse assortment of BRAF mutations. In addition, in melanoma patients whose tumors were resistant to BRAF/MEK dual inhibition, ulixertinib is shown to exert anticancer effects[31]. Due to the success of ulixertinib thus far, the inhibitor is being investigated in combination with other inhibitors, such as BRAF inhibitor dabrafenib[31].
Despite the initial success of ulixertinib, challenges remain with ERK inhibitors. A 2018 study by Jaiswal et al.[32]conveyed that ERK inhibitors, when used as a single agent, are likely to confer drug-acquired resistance. The researchers looked at five different ATP-competitive ERK inhibitors, including ulixertinib, to determine the mechanism behind acquired resistance to ERK inhibition. Upon treatment with ERK inhibitors, RAS-/RAF-mutant cell lines acquired several mutations that affect the binding of ERK inhibitors to their target, preventing the hindrance of ERK1/2 activity[32]. The authors also displayed the novel finding that ERK2 amplification is a mechanism of resistance to ERK inhibitors[32]. Hence, due to the diverse mechanisms of resistance to ERK inhibition, ERK inhibitors are being looked at in combination with other inhibitors of the ERK/MAPK pathway. Jasiwal et al.[32]suggested that ERK inhibitors in combination with MEK inhibitors may prove advantageous in treating malignancies with acquired resistance to RAF/MEK inhibitors. In addition, RTKs and components of the PI3K/AKT/mTOR pathway were identified as mediators of acquired resistance. As such, combination therapy involving ERK inhibitors with either inhibitors of the PI3k/AKT/mTOR pathway or RTK inhibitors, such as those that target members of the HER family, may prevent the development of resistance[32]. Overall, the use of ERK inhibitors as part of combination therapy is promising for the treatment of cancers with mutations in the ERK/MAPK pathway.
HER2/NEU/ERBB2
The RTK EGFR consists of four members, also known as the human epidermal growth factor receptor (HER) or ErbB family. These receptors include HER1 (also called ErbB1 or EGFR), HER2 (also called ErbB2), HER3 (ErbB3), and HER4 (ErbB4). Under normal conditions, the members of this family of RTKs are known to have important roles in development. Like other RTKs, the HER/ErbB family of receptors are activated by binding to ligands on their extracellular domains, which leads them in turn to form dimers with each other or with other HER/ErbB family members. Upon dimerization, intracellular signaling pathways are activated, leading to a response to the signal. HER2/ErbB2 functions differently from the other members because it does not require a ligand to form a dimer. Although it does not directly bind any ligands, HER2 is activated by homodimerization or heterodimerization with HER1, HER3, or HER4[33,34][Figure 2].

Figure 2. Her2 signaling: HER2 is a member of the HER/EGFR family of RTKs. HER2 signaling has important roles in regulating normal cell function. However, its dysregulation is often implicated in cancer. HER2 can homodimerize with itself or heterodimerize with the other members of the HER family. Overexpression of HER2 can lead to the overactivation of the MAPK/ERK and PI3K/AKT/mTOR pathways, leading to uncontrolled cell growth and evasion of apoptosis. HER: human epidermal growth factor receptor; EGFR: epidermal growth factor receptor; RTK: receptor tyrosine kinase; MAPK: mitogen-activated protein kinase; ERK: extracellular signal-regulated kinases; PI3K: phosphoinositide 3-kinase; AKT: protein kinase B; mTOR: mammalian target of rapamycin
The signaling pathways regulated by the HER receptors have important roles in responding to extracellular signals and in turn regulating normal cell function. Improper regulation of the HER receptors, especially HER1 and HER2, can be associated with cancer. In healthy cells, HER2 activation triggers signaling pathways that control normal cell growth, differentiation, motility, and adhesion. Overexpression of HER2 leads to the over-activation of downstream pathways, including the PI3K/Akt/mTOR and MAPK/ERK signaling cascades, which are known to be involved in malignant transformation[35]. The overexpression of HER2 signaling is shown to promote the loss of cell adhesion and polarity, defining features of epithelial cancers[36,37]. As such, HER2 dysregulation is associated with ovarian cancer, gastric cancer, and perhaps most notably, breast cancer.
HER2 in breast cancer
Breast cancer is the most common type of cancer among women in the United States, and 20-30% of breast cancers are thought to be HER2-positive. Amplification of the HER2 gene in breast cancer is associated with invasiveness, large tumor size, and late clinical stage. HER2 overexpression is also associated with higher rates of recurrence and shorter rates of survival in patients with breast cancer[38,39]. Once activated, HER2 is shown to recruit proto-oncogene Src. The constitutive activation of the Src kinase by the overexpression of HER2 has a role in tumor progression and metastasis and is implicated in breast cancer[40]. Moreover, one way in which HER2 promotes metastasis is by its upregulation of the chemokine receptor CXCR4. HER2-mediated CXCR4 expression in breast cancer is shown to be instrumental in the movement of malignant cells to specific tissues, which contributes to poor survival rates[41,42]. In addition, HER2 overexpression in epithelial breast cancer cells is shown to deregulate the G1/S cell cycle checkpoint. HER2 exerts its effects on G1/S control by upregulating cyclins D1 and E and cyclin-dependent kinase CDK6, and by enhancing the degradation of cyclin-dependent kinase inhibitor p27Kip1. These changes in cell cycle progression contribute to the enhanced signaling of the PI3K/Akt and MAPK pathways[43].
In addition to CXCR4, HER2 interacts with several other tumor markers. For instance, estrogen receptor (ER) and progesterone receptor (PR) are important for the prognosis and diagnosis of breast cancer. The overexpression of ER and PR in breast cancer is associated with a better clinical outcome when compared to tumors that overexpress HER2. Notably, ER and PR expression are inversely correlated to HER2 expression. There are several explanations for this occurrence. For example, the hyperactivation of the MAPK pathway in response to HER2 overexpression contributes to the downregulation of ER. Hence, the overexpression of HER2 leads to decreased ER and PR expression and promotes oncogenesis[33,44]. Likewise, breast cancers with HER2 amplification have been shown to overexpress the angiogenic growth factor VEGF. The HER2-mediated upregulation of VEGF expression is associated with aggressiveness and high rates of recurrence in HER2-positive breast cancers[45]. Lastly, recent evidence shows that HER2 disrupts the function of the tumor suppressor E-cadherin. E-cadherin is a transmembrane protein that mediates epithelial cell adhesion and tissue formation. Cell-cell adhesion by E-cadherin is maintained through its interaction with β-catenin and the cytoskeleton. HER2 disrupts the E-cadherin/β-catenin complex by binding and phosphorylating β-catenin. This blocks the interaction between E-cadherin and β-catenin, resulting in the loss of E-cadherin expression. As such, the loss of E-cadherin is found in over half of invasive lobular carcinomas at both the primary tumor and metastatic sites[46-48]. Accordingly, HER2 overexpression is shown to have a prominent role in the invasiveness of breast tumors and serves as a critical drug target.
Targeting HER2 in breast cancer
The advent of anti-HER2 therapies, along with the increase of early cancer screenings, have led to significant improvements in the survival rates of HER2-positive breast cancer over the past two decades. Anti-HER2 therapies, such as the monoclonal antibody trastuzumab (Herceptin) and the tyrosine kinase inhibitor lapatinib (Tykerb), have shown to be clinically successful in treating HER2-positive breast cancers. Nonetheless, a significant proportion of HER2-positive breast cancer patients who were treated with trastuzumab relapse and develop metastatic disease. This indicates that tumors possess or develop resistance to anti-HER2 therapies[43]. In order to discern the many mechanisms of intrinsic and acquired resistance of HER2-positive tumors, it is important to understand how trastuzumab works to inhibit HER2. Trastuzumab is shown to downregulate HER2 by binding its extracellular domain, and to block HER2-HER3 dimerization[49,50]. Consequently, trastuzumab exercises several anti-tumor effects in HER2-overexpressing cells by downregulating PI3K/AKT pathway signaling along with downregulating mediators of cell cycle progression, such as cyclin D1, and upregulating the p27 cell cycle inhibitor[51,52].
It is critical to understand the various mechanisms behind trastuzumab resistance in order to develop novel anti-HER2 therapies. One way in which HER2-positive cancer cells become resistant to trastuzumab is through the constitutive activation of the PI3K/Akt/mTOR signaling cascade. Loss of function mutations in tumor suppressor PTEN and activating mutations in PI3K lead to aggressive cell growth. These alterations in the PI3K/Akt pathway are present in patients who have been treated with trastuzumab and are thought to be associated with drug resistance[51]. Another leading way that HER2 overexpressing tumor cells escape anti-HER2 therapies is through the expression of p95HER2. Approximately 30% of HER2-positive breast cancers express the p95HER2 fragment, which is a truncated form of HER2 that is especially oncogenic, as it spontaneously homodimerizes into a constitutively active form. Tumors with p95HER2 fragments are resistant to trastuzumab because they lack the extracellular domain and the binding site for trastuzumab[53,54]. Unlike trastuzumab, HER2 RTK inhibitor lapatinib can inhibit p95HER2 along with HER2, as it binds the ATP active site present in both proteins. This inhibition blocks phosphorylation and inhibits downstream signaling[55].
Although lapatinib has some advantages, cancer cells can still develop resistance. When HER2 signaling is suppressed by lapatinib and trastuzumab alike, alternative survival mechanisms can be activated. Even though HER2 and p95HER2 are shown to be inhibited by lapatinib, the suppression of HER2 signaling can lead to the upregulation of other HER family members and RTKs, such as the fibroblast growth factor receptor-2 and insulin-like growth factor-1. Alternately, lapatinib resistance can be attributed to the hyperactivation of the PI3K pathway by the activation of Src kinases through insulin receptor substrate 4. Recently, another anti-HER2 agent, neratinib, was evaluated in clinical trials. Unlike lapatinib, neratinib is an irreversible pan-HER inhibitor[56]. A recent phase III clinical trial compared the effects of neratinib versus lapatinib in combination with capecitabine, an antimetabolite, in patients with HER2-positive metastatic breast cancer. The results indicate that neratinib + capecitabine significantly improved progression-free survival over lapatinib + capecitabine. These results offer a promising outlook on the use of neratinib combination therapy in the treatment of aggressive HER2-positive breast cancers[57].
Trastuzumab and lapatinib-resistant cells continue to depend on HER2 to evade drug treatment. One way in which these late stage drug-resistant cancer cells continue to depend on HER2 is by expressing MUC4, a membrane-associated mucin that masks HER2. This is shown to prevent the binding of trastuzumab to HER2 and to maintain HER2 phosphorylation. Knockdown of MUC4 with RNA interference (RNAi) led to an increased binding of HER2 to trastuzumab[58]. Studies have also been designed to investigate the use of RNA interference directly against HER2. The results of a recent study demonstrate that RNAi of HER2 by siRNA has antiproliferative effects and decreases HER2 mRNA and protein levels in vitro[59,60]. In addition, another study looked at the effects of siRNA on BT474 cells (ductal carcinoma). The authors report that siRNA treated BT474 cells do not acquire resistance against the treatment. This work has not yet been translated to in vivo studies, but these preliminary results demonstrate that siRNA poses as a promising option for patients with drug-resistant HER2-positive breast cancer[61].
PI3K/AKT/MTOR PATHWAY
The mammalian target of rapamycin (mTOR) pathway is another example of an important signaling protein that can operate activated downstream to RTKs [Figure 3]. mTOR is a serine/threonine kinase. Together with other proteins, it forms two different complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 is activated by RTKs as well as by other types of stimuli. It is part of a signaling pathway that includes phosphoinositide 3-kinase (PI3K) and AKT, which are implicated in cell survival [Figure 3]. The mTOR pathway is activated in a wide variety of solid tumors and is one of the main causes of resistance to anti-tumor therapies. The mTORC1 signaling cascade is involved in growth, proliferation, motility, metabolism, and immune response regulation. Hyperactivation of the PI3K/AKT/mTOR pathway leads to genomic instability and uncontrolled proliferation of cancer cells. Thus, due to its role in tumorigenesis and drug resistance, the PI3K/AKT/mTOR cascade is a prominent therapeutic target for several types of cancers[62].
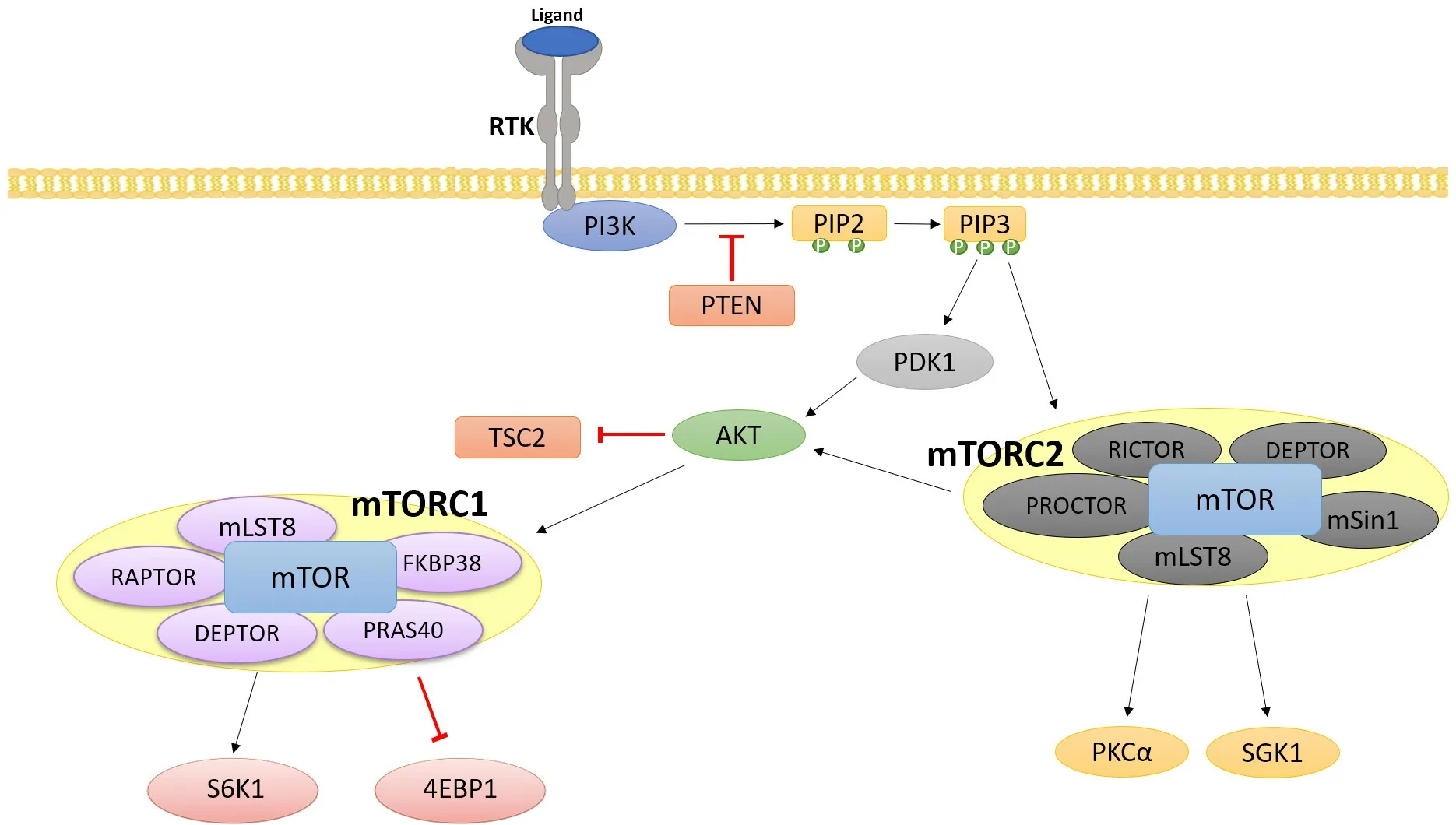
Figure 3. PI3K/AKT/mTOR signaling cascade: The PI3K/AKT/mTOR pathway is activated through RTK signaling, and hyperactivation of this pathway is implicated in several cancers. The main effectors of this pathway, mTORC1 and mTORC2, are major regulators of translation, cell metabolism, and cell proliferation. RTK: receptor tyrosine kinase; PI3K: phosphoinositide 3-kinase; AKT: protein kinase B; mTOR: mammalian target of rapamycin
PI3K is an important upstream activator of mTORC1. The PI3K family is a group of lipid kinases that regulates a broad array of cellular functions in signaling and metabolism. Of the 3 classes of PI3Ks, the class I PI3Ks are most often linked to oncogenesis[63]. The activation of G-coupled protein receptors and autophosphorylation of tyrosine residues of RTKs recruit PI3K to the membrane and activate its catalytic subunit. PI3K activation leads to the phosphorylation of PIP2 to produce secondary messenger phosphatidylinositol (3, 4, 5)- trisphosphate (PIP3). PIP3 amplifies the initial signal from PI3K and subsequently recruits 3’-phosphoinositide-dependent kinase-1 (PDK1) and protein kinase B/Akt to the membrane. PDK1 is responsible for the activation of Akt, and Akt in turn regulates several downstream effectors. Notably, Akt activates the Forkhead Box O proteins and the mTOR pathway[64].
The mTOR pathway is a major regulator of translation and consists of two complexes, mTORC1 and mTORC2. AKT can both directly and indirectly activate the mTOR pathway. AKT is shown to activate the mTORC1 complex through the phosphorylation and subsequent inactivation of two negative regulators of the mTOR pathway - tuberous sclerosis complex 2 and PRAS40 (proline-rich Akt substrate of 40kDa). In mTORC1, mTOR associates with Raptor, which regulates mTOR activity and recruits mTORC1 substrates. mTORC1 regulates ribosomal proteins S6K and 4E-BP1, which have roles in regulating protein synthesis and metabolism[65][Figure 3]. The phosphorylation and activation of S6K by mTORC1 leads to the phosphorylation of S6, which advances mRNA translation and further promotes the synthesis of rRNAs, tRNAs, and transcription factors. On the other hand, mTORC1 inactivates 4E-BP1 which plays a role in the inhibition of translation. Hence, when 4E-BP1 is phosphorylated by mTORC1, translation initiation is promoted[66]. Unlike mTORC1, less is known about the activation and regulation of mTORC2. However, evidence shows that mTORC2 is activated by PIP3 and growth signals that can promote cell proliferation and survival [Figure 3]. Similar to the role that Raptor has in mTORC1 regulation, Rictor associates with mTORC2 and is important in stabilizing and monitoring mTORC2 activity. Additionally, mTORC2 is shown to regulate small GTPases and members of the AGC kinase family, which includes AKT, PKC, and SGK1 (protein kinase C)[64,67]. As such, the dysregulation of both mTORC1 and mTORC2 have been shown to exert oncogenic effects.
All known major effectors of the PI3K/AKT/mTOR pathway are frequently mutated in cancer. As previously mentioned, PI3K depends upon RTKs for its activation. Because the majority of PI3K is inactive in the cytoplasm, even miniscule changes in RTK activity can lead to overactivation of PI3K. For instance, mutations that lead to increased activation of EGFR (ERBB1) and HER2 are associated with cancers with aberrant PI3K pathway signaling[68]. In addition, activating mutations in the platelet derived growth factor (PDGFRα) RTKs are found to be present in several cancers, including gastrointestinal stromal tumors and mammary carcinomas[69,70]. Further, activating missense mutations in PIK3CA, the gene that encodes for the p110α catalytic subunit of PI3K, are found in a wide variety of cancer types. Tumor suppressor phosphatase and tensin homolog (PTEN) regulates and represses PI3K activity by the dephosphorylation of secondary messenger PIP3. PTEN, however, is found to be mutated in several tumors which results in the constitutive activation of PI3K. Activating mutations in PI3KCA and inactivating mutations in PTEN are often found to coexist in many cancers, such as breast cancer and colon cancer. Amplification of AKT is also seen in cancer as a result of mutations in the AKT1 gene[71,72]. Lastly, there are various cancer-related mutations in the MTOR gene. These mutations affect the activity and assembly of both mTORC1 and mTORC2[73].
Pl3K inhibitors
There are three main classes of PI3K inhibitors - pan-PI3K inhibitors, isoform specific PI3K inhibitors, and dual PI3K/mTOR inhibitors. Several pan-class I PI3K inhibitors are currently in clinical development[74]. Pan-class I PI3K inhibitors exert inhibitory effects against each isoform of PI3KCA, the gene that encodes for p110α, the catalytic domain of PI3K. Buparlisib, for instance, binds to the ATP binding site of the lipid domain of PI3K and exhibits potent activity in cancers with PI3KCA activating mutations that are found in a wide range of human cancers. In addition, copanlisib, another pan-PI3K inhibitor, also shows significant anti-tumor activity in cancers with PI3KCA mutations and in cancers with HER2 overexpression. Pan-class I PI3K inhibitors have shown anticancer effects when administered as single agents. Nonetheless, there are multiple side effects that limit the efficacy of class-I inhibitors, such as hyperglycemia and fatigue[75-77]. Further, isoform specific PI3K inhibitors have advantages over pan-PI3K inhibitors because they only bind to one isoform of PI3K. There are four class-I PI3K isoforms (PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ), and each isoform plays non-redundant roles in particular tumor types. Hence, by targeting one isoform, undesirable side effects can be limited[78].
As previously mentioned, mutations in the PI3KCA gene are recurrent in solid tumors. There are several selective and highly specific inhibitors that target the PI3Kα isoform, which is composed of the p110α catalytic subunit encoded by PI3KCA. Preclinical studies have shown that HER2- and KRAS-driven tumors rely on PI3Kα for tumor survival. Hence, the inhibition of this isoform by selective PI3Kα inhibitors acts as a promising therapeutic target. These inhibitors have been shown to have potency against wild-type PI3Kα and E545K and H1047R gain-of-function mutants. Such inhibitors, such as NVP-BYL719, are shown to block the phosphorylation of AKT, which is highly dependent on p110α activity. In addition, isoformspecific PI3Kα inhibitors are shown to decrease glucose consumption and blood supply to cancer cells, contributing to their anti-tumor effects. As such, further studies are being carried out to determine whether using isoform-specific PI3Kα inhibitors in combination with other agents enhances tumor regression[79,80].
In addition to PI3Kα, targeting the PI3Kδ isoform may be beneficial for certain cancers. PI3Kδ is largely seen in leukocytes and serves as an important target for cancer and immune-related diseases. As it is downstream to several B-cell receptors, PI3Kδ plays an integral role in B-cell signaling and is seen in B-cell related malignancies. Mutations in the PI3KD gene have not been found in patients with chronic lymphocytic leukemia, but previous studies report that increased PI3K activity is highly dependent on PI3Kδ isoform as observed in these cancers. PI3Kδ selective inhibitor CAL101 blocks constitutive PI3K signaling that confers cancer cell survival, such as the phosphorylation of AKT and ERK1/2. Notably, CAL101 has also been demonstrated to reduce the levels of circulating chemokines CCL3, CCL4, and CXCL13, which are associated with tumor metastasis and migration[79,81]. Moreover, PI3Kβ-selective inhibitors are promising for the treatment of a group of solid tumors. PI3Kβ inhibitors were shown to inhibit tumor cell growth in PTEN-null advanced solid tumors. Nevertheless, prolonged treatment of PTEN-deficient cancers with PI3Kβ inhibitors may cause tumor cells to shift their dependency from the PI3Kβ to the PI3Kα isoform[82].
mTOR inhibitors
mTOR was originally identified to be a target for the anti-fungal agent rapamycin[83]. The dysregulation of mTOR is shown to promote cancer growth and metastasis and mTOR thus serves as an important therapeutic target. mTOR regulates basic physiological processes and is frequently mutated in cancers. mTOR is the central component of both the mTORC1 and mTORC2 complexes. Both complexes differ in their components, substrate specificities, and subcellular localization. Aberrant mTOR signaling can be activated in various ways in cancer. First, mutations in the mTOR gene or in the genes that encode components of the mTORC1/2 complexes can lead to constitutive mTOR signaling. In addition, gain-offunction mutations in upstream oncogenes or loss-of-function mutations in upstream tumor suppressors contribute to unregulated mTOR signaling[84]. Rapamycin, the compound from which mTOR gets its name, was the first agent that was discovered to bind mTOR. It was shown to bind and inhibit mTORC1, but due to its low solubility and poor pharmacokinetics, it is unsuitable for treating human cancers. As a result, several rapamycin analogs, “rapalogs”, have been discovered. The rapalogs everolimus and temsirolimus are prescribed to treat advanced renal cell carcinoma and breast cancer. It is evident that everolimus and temsirolimus exert tumor-suppressive effects in vivo, but the benefits of such agents in the clinic is only moderate. The limited clinical success of rapalogs is a result of the incomplete inhibition of mTOR and the development of acquired resistance in tumors. Nonetheless, clinical trials to determine the efficacy and breadth of rapalogs in different cancers is ongoing[85].
In order to more completely suppress mTOR, ATP-competitive mTOR inhibitors have been developed to target both mTORC1 and mTORC2. Many of these ATP-competitive inhibitors target both mTOR and PI3K. In various cases, the dual inhibition of PI3K and mTOR is seen to be more effective than targeting either protein alone. For instance, in a recent study the effectiveness of the ATP-competitive PI3K/mTOR dual inhibitor BEZ235 in the treatment of drug-resistant gastric cancers was assessed. BEZ235 inhibits multiple class I PI3K isoforms and mTORC1/2 activity. Unlike mTOR inhibitors alone, BEZ235 was shown to decrease AKT activity in vitro, contributing to its potent anticancer effects. However, it is important to note that although BEZ235 has significant anti-tumor activity, it is shown to enhance ERK activity through mTORC2. The use of BEZ235 in combination with MEK inhibitors is seen to prevent ERK overactivation and to significantly inhibit cell growth[86,87]. Similarly, another study reported the use of a newly developed PI3K/mTOR dual inhibitor, CMG002, in chemoresistant ovarian cancer. CMG002 was shown to suppress cancer cell proliferation and to induce apoptosis and G1 arrest in drug-resistant ovarian cancer cells[88]. As such, both BEZ235 and CMG002 as single agents serve as promising strategies in cancer treatment. Further work is being done to study the effects of dual PI3K/mTOR inhibitors in combination with inhibitors that target the Ras/Raf/MEK/ERK pathway.
THE PAK FAMILY
The Pak family of protein kinases are important signaling proteins implicated in many cellular functions including cell proliferation, migration, and cytoskeletal organization[89]. They can mediate a response to RTKs and other activators. They can lead to activation of MAP kinases and other signaling pathways. They were first identified as protein kinases that bind and are activated by the Rho GTPases Rac and Cdc42 (Fig. 4). The Pak family consists of 6 members. These include group A (Paks 1, 2, and 3) and group B (Paks 4, 5, and 6)[89]. The Paks have several different domains, including a GTPase binding domain (GBD), a short sequence which can bind to the Rho GTPases Cdc42 and Rac, and a serine/threonine kinase domain.
The group A and group B Paks have approximately 50% amino acid identity in the GBD and kinase domains, but differ throughout their other domains[89]. Among the Pak family members, the Pak1 and Pak4 genes are located on chromosomal regions that are frequently amplified in cancer[90]. Increased Pak4 levels are associated with oncogenic transformation[91,92]and occur in a number of cancers[93-96]. The wildtype Pak4 gene was shown to be amplified in a panel of pancreatic cancer samples, in pancreatic ductal adenocarcinomas[95,97,98], in squamous cell carcinomas[99], and in high grade serous and endometrioid ovarian tumors[100]. In ovarian cancer cell lines, siRNA studies have shown that Pak4 is one of the genes whose amplification is critical for cellular viability[100]. High levels of Pak4 are found across all subtypes of breast cancer including ER+, Her2+, and triple negative[92,101-105]. Thus, Pak4 could serve as a more specific target for breast cancer therapy.
Pak4 has several cellular functions that may explain why it is so tightly linked with cancer. For example, Pak4 leads to cytoskeletal changes. This may be directly linked with the control of cell shape, which is often disrupted in cancer, and with cell migration[106], a critical function for cancer cells, particularly during metastasis [Figure 4]. Pak4 has also been shown to have key roles in promoting cell survival[107-109]. It can promote cell survival and inhibit apoptosis in response to several different stimuli in both fibroblasts and epithelial cells, including serum withdrawal, or cytokine stimulation, and it can do so by a combination of different mechanisms, which involve both kinase dependent and kinase independent functions[107-109]. Pak4 can also promote cell proliferation, another important aspect of tumorigenesis, and in some cells, this is associated with a prolonged activation of the ERK MAP kinase pathway[101].
Due to the strong link of Pak4 to cancer, several anti-Pak4 compounds have been developed[110-114]. One of these compounds is KPT-9274. Unlike other Pak4 inhibitors that block PAK4 catalytic activity, KPT-9274 reduces Pak4 protein levels. This may be important because Pak4 has been shown to have kinase independent functions[96,108]. For example, a kinase dead Pak4 mutant can inhibit caspase 8 activity and consequently increase cell survival, as well as a wild-type Pak4. Pak4 can also regulate adhesion turnover and thereby promote migration, by a mechanism that is independent of its kinase activity[92,113]. KPT-9274 could thus be more effective in antagonizing Pak4 functions in cancer compared with inhibitors that specifically block kinase activity. KPT-9274 has shown promise for the treatment of breast cancer, renal cancer, pancreatic cancer, and others both in vitro and in vivo and is now in a phase I clinical trial for patients with advanced solid malignancies and non-Hodgkin’s lymphoma (NCT02702492)[114-119]. Although KPT-9274 was identified as a Pak4 allosteric inhibitor, it also inhibits the metabolic enzyme nicotinamide phosphoribosyltransferase (NAMPT) by an undefined mechanism[115]. NAMPT is the rate limiting enzyme for the production of nicotinamide adenine dinucleotide (NAD) via the salvage pathway[120]. NAD is an important cofactor for redox reactions in metabolic pathways and its levels are tightly controlled by mechanisms that remain poorly understood. Whether Pak4 is linked to NAMPT, and how dual inhibition of Pak4 and NAMPT by KPT-9274 leads to growth inhibition of cancer cells warrant further investigation.
Recently, KPT-9274 was not only shown to lead to inhibition of Pak4 and NAMPT in triple negative breast cancer cells, but also inhibition of Rictor[121]. Rictor is an important component of mTORC2, Conversely, mouse mammary epithelial cells overexpressing Pak4 had an increase in Rictor activity[121,122]. AKT phosphorylation at Ser473 was dramatically decreased in response to KPT-9274 treatment, also a hallmark of mTORC2 activation. These findings reveal that prolonged inhibition of Pak4 by KPT-9274 inhibits mTORC2 activity, and mTORC1 activity was subsequently also found to be decreased. These results suggest that the anti-proliferative effects of KPT-9274 could be mediated by inhibition of mTOR signaling. They also raise the possibility that the PAK4 pathway is linked to the mTOR pathway, possibly via phosphorylation of a component of mTORC2. These results suggest that the simultaneous inhibition of both Pak4 and mTOR may be an effective way to block the growth of breast cancer cells, and this may have implications in other types of cancer as well.

Figure 4. The role of PAK4 in cell signaling: external signals initiate the activation of small GTPases Cdc42 and Rac, which in turn bind to and activate PAK4. PAK4 has roles in cytoskeletal organization, motility, adhesion, and the regulation of gene expression. mRNA and protein levels of PAK4 are often seen to be overexpressed in several types of cancer, such as breast cancer and pancreatic cancer. GBD: GTPase binding domain
CONCLUSION
This review has highlighted some of the RTK regulated pathways that are important for cancer and for drug development. Several important RTK regulated pathways have been discussed. It is important to remember, however, that there is are many more RTKs than we have discussed here. In fact there are at least 58 unique RTKs, which fall into at least 20 families[123]. Several diあerent drugs have been developed against proteins that are improperly regulated in RTK pathways. Even more are currently in development or under clinical trials. Some of these have had promising outcomes. It is important to remember, however, that serious challenges remain ahead. One issue is the development of resistance to drugs that were initially successful[2]. To address this, current and future studies need to address the reason for this resistance, and the development of new drugs that will be effective for resistant cancers[124]. Another challenge is the development of combination treatments. Single agent treatments are often not sufficient for eあective treatments, but drug combinations can be more eあective. One promising area of study is the combination of protein kinases with immune complex inhibitors, in order to combine immunomodulation with inhibition of RTK signaling pathways. Finally, identifying which treatments will be most eあective in each individual patient will contribute to a long-sought goal of developing individualized cancer treatments for each patient.
DECLARATIONS
Acknowledgments
The authors are grateful to Busch Biomedical Grant and the New Jersey Health Foundation for providing support for the lab.
Authors’ contributions
Did the majority of the writing for this manuscript: Cordover E
Helped with the planning of the article, some of the writing, and editing: Minden A
Availability of data and materials
Not applicable.
Financial support and sponsorship
Financial support for the lab and for Emma Cordover’s work was from the Busch Biomedical Grant, the New Jersey Health Foundation Grant, and the Aresty Research Center.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2020.
