进行性家族性肝内胆汁淤积症3型一家系2例报告
2020-07-21李爱芹徐志强王福川王丽旻闫建国曹丽丽
李爱芹,董 漪,徐志强,王福川,王丽旻,闫建国,曹丽丽,王 璞,张 敏
中国人民解放军总医院第五医学中心 青少年肝病诊疗与研究中心,北京 100039
进行性家族性肝内胆汁淤积症(progressive familial intrahepatic cholestasis,PFIC)是一组婴儿或儿童期起病,以严重肝内胆汁淤积及皮肤瘙痒为主要特征,在儿童期或者青春期可因肝衰竭而导致死亡的罕见常染色体隐性遗传性疾病。目前确切发病率尚无报道,估计为1/50 000~1/100 000[1]。PFIC共分6型,是我国儿童慢性胆汁淤积的重要原因之一。其中,1、2、4、5、6型的血清Y-GGT特征性降低或正常,而由ABCB4基因突变导致的PFIC 3型(PFIC-3)GGT升高,肝组织病理表现为明显的小胆管增生和肝纤维化,与非PFIC导致的胆汁淤积性肝病不易区分,容易造成误诊漏诊。本文将一家系中兄弟2人临床表现为胆汁淤积、进行性肝纤维化,历时多年诊断不明,最终基因检测确诊为PFIC-3的病例报道如下。
1 病例资料
病例1:患者男性,15岁,因“发现肝功异常2年”于2009年8月25日入住本院。2007年8月其母亲发现患儿颜面部毛细血管显现,当地医院查肝功能异常,甲、乙、丙、戊病毒学检查均阴性,铜蓝蛋白(CER)正常,B超提示肝脾大。予保肝降酶药治疗后效果不佳而入本院。入院查体:生命体征平稳,全身皮肤粗糙,可见散在抓痕,皮肤巩膜无黄染,颜面部可见毛细血管扩张,肝掌阳性。心肺无异常,腹平软,肝右肋下6 cm可触及,剑突下3 cm可触及,质硬、边锐,无触痛,脾脏左肋下6 cm触及(平脐),质中,边钝,无触痛。移动性浊音阴性,双下肢不肿。辅助检查:WBC 4.51×109/L、Hb 109.0 g/L、PLT 83.0×109/L、白蛋白34 g/L、球蛋白25 g/L、TBil 18.5 μmol/L、DBil 9.9 μmol/L、ALT 104 U/L、AST 190 U/L、总胆汁酸(TBA)30 μmol/L、胆碱酯酶(ChE)2932 U/L、PT 13.0 s、凝血酶原活动度(PTA)82.0%、CER 0.42 g/L。尿铜102.2 μg/24 h(参考值15~30 μg/24 h),予青霉胺试验后尿铜378 μg/24 h。铜氧化酶吸光度0.26。双角膜K-F环阴性。颅脑MRI未见异常。骨髓穿刺结果未见明显异常。腹部超声示:轻度脂肪肝、脾大、脾静脉扩张。肝脏病理:可见肝小叶结构紊乱,假小叶形成,肝细胞弥漫性水样变性,轻度脂肪变性,易见呈假性腺样排列的肝细胞,部分胞浆嗜酸性变,汇管区纤维性扩大,少量炎细胞浸润,增生纤维组织较宽大且致密,小胆管轻度增生,轻度界面炎;铜染色:阳性。考虑肝硬化,活动期,不除外Wilson病所致。曾于外院行ATP7B基因学检测,提示存在1个变异位点杂合子,但未确诊,依据病理诊断为 “肝豆状核变性”,予联苯双酯、硫酸锌片、二巯丁二酸胶囊、复方氨基酸等保肝、排铜治疗,但患儿瘙痒无改善,每半年~1年随访复查肝功指标如表1,肝功能渐差,影像学检查显示肝硬化、脾大逐渐进展。2013年1月腹部CT提示肝硬化、脾大、脾肾分流,肝S6占位性病变,不除外肝癌。家属拒绝治疗,定期观察,占位未见明显增大。2015年12月因其弟弟(患者2)婴儿期出现肝功能异常,病理明显纤维化考虑存在家系遗传代谢病,强烈建议再次行基因检查。遂2015年8月行兄弟二人基因检查。
病例2:患者男性,4岁,因“发现肝功能异常3个月”于2015年8月13日入住本院。缘于2015年5月因“支气管肺炎”在当地医院检查发现ALT 195 U/L、HBsAg阴性,为进一步诊治入住本院。入院查体:全身皮肤巩膜无黄染,肝掌阴性,未见蜘蛛痣。心肺检查无异常,双下肢不肿。辅助检查:PLT 151.00×109/L、WBC 14.61×109/L、Hb 125.00 g/L、血氨31.00 μmol/L、PT 10.3 s、PTA 97.9%、ALT 111 U/L、肌酸激酶320 U/L、AST 127 U/L、ALP 414 U/L、GGT 126 U/L、铜蓝蛋白0.43 g/L。腹部超声示:肝脏增大。肝脏病理:肝小叶结构紊乱,早期假小叶结构形成。肝细胞假腺样排列易见,弥漫性水样变性,区域性气球样变,少数肝细胞脂肪变性,散在点灶状坏死,凋亡小体易见;窦周炎及窦周纤维化可见;汇管区明显扩大,纤维组织增生,纤维间隔易见,大量混合性炎细胞浸润,嗜酸性粒细胞易见,轻度界面炎。小胆管增生。提示慢性肝炎,病变程度相当于G2S4,结合基因学检查除外肝豆状核变性,并不完全除外重叠非嗜肝病毒、药物等因素。外院病理会诊:肝内轻度铜沉积,慢性淤胆伴肝纤维化S3,符合Wilson病。血、尿筛查无明显异常。由于与其兄(病例1)症状相似,考虑遗传代谢病可能性大,故兄弟同时于2015年12月行基因检测。
基因检测(北京金准基因科技有限责任公司)结果(图1):2个患者在ABCB4基因外显子区域发现3处相同的杂合突变点:c.2570C>T(胞嘧啶>胸腺嘧啶),chr7:87046740,导致p.T857I(苏氨酸>异亮氨酸);c.2212A>T(腺嘌呤>胸腺嘧啶),chr7:87051541,导致p.I738F(异亮氨酸>苯丙氨酸);c.1694C>G(胞嘧啶>鸟嘌呤),chr7:87069020,导致p.T565R(苏氨酸>精氨酸)。进一步通过 Sanger 测序验证,确认c.2570C>T来自于母亲,另外两个杂合突变点来自于其父亲。HGMDpro数据库报道情况:均未见报道。兄弟俩有相似的临床症状,父母正常,以此推断在此家系中遵循隐性遗传规律,理论上此三处杂合突变仍为复合杂合突变,若母源突变为致病性突变,其父源的俩个突变至少有一个致病性突变,理论上有致病的可能。软件预测结果如图2,有致病风险,诊断为PFIC-3。
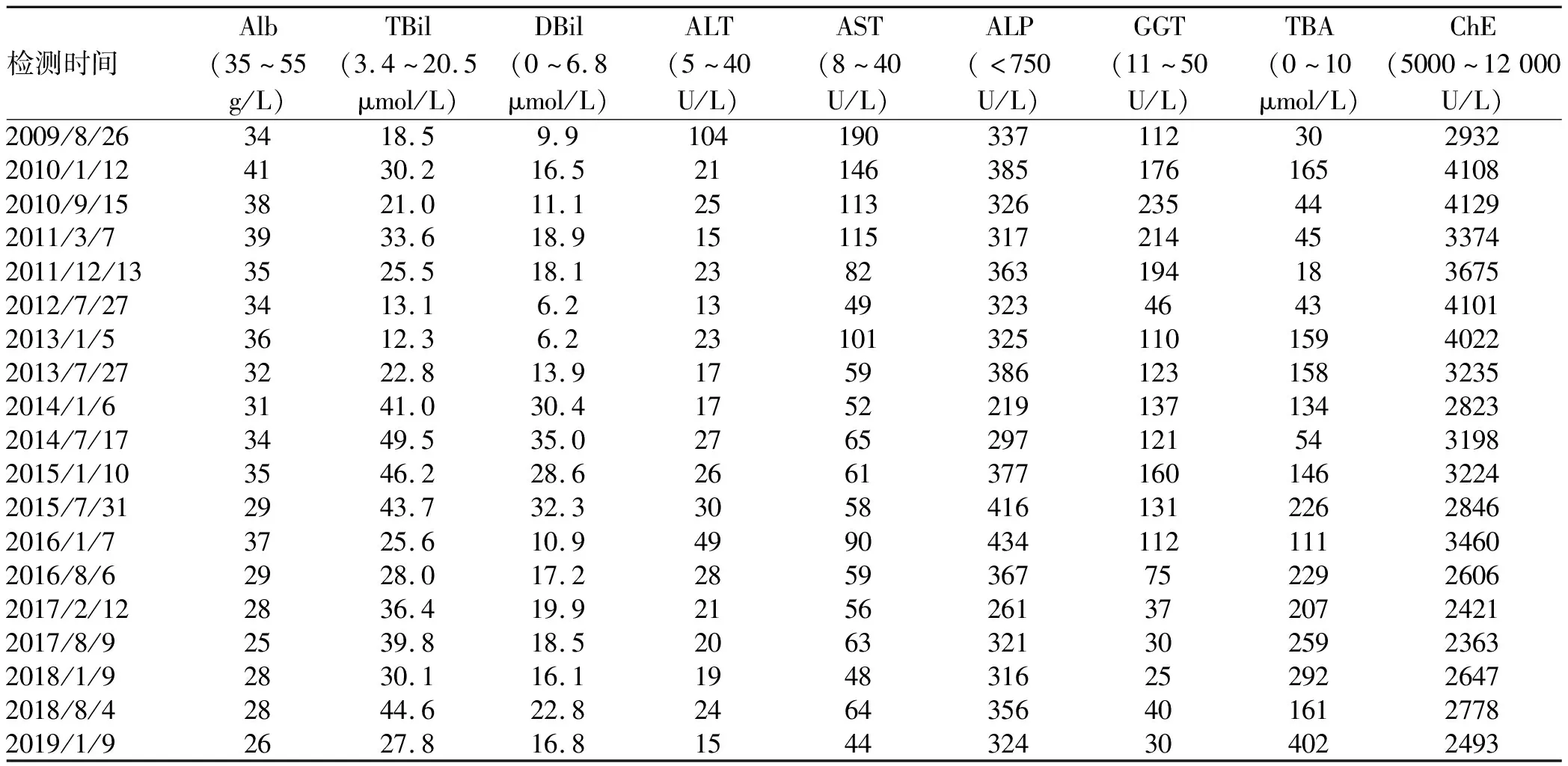
表1 病例1历次生化检查结果
2016年2月底基因结果回报,兄弟二人诊断为ABCB4 基因突变导致的PFIC-3。哥哥停用二巯丁二酸、硫酸锌,予口服联苯双酯、复方氨基酸、熊去氧胆酸等对症治疗,2019年1月影像学检查仍无明显改善,发展至肝硬化失代偿期,Alb、CHE、TBA等生化指标仍异常,但GGT下降至正常(表1)。弟弟确诊为 PFIC-3后,经熊去氧胆酸、双环醇维持治疗,随访3年疾病无明显进展,Alb正常,转氨酶、GGT、TBA均降至正常(表2),但仍有皮肤瘙痒,目前仍在定期随访中。
2 讨论
根据致病基因不同,可将PFIC分为1~6型,PFIC-3是PFIC的一种亚型[2]。大多数PFIC患者均有血清TBA和转氨酶升高,多数伴有血清胆红素及ALP水平升高,胆汁中初级胆汁酸水平降低,各型PFIC除PFIC-3患者外,血清GGT均基本正常甚至偏低,籍此可以区别于其他原因引起的胆汁淤积性肝病。但PFIC-3的GGT水平是升高的,因此与其他原因引起的胆汁淤积性肝病不易区分[3]。
PFIC-3即ABCB4 缺陷病,是编码多耐药蛋白3(multi-drug resistance-3-p-glycoprotein,MDR3)的基因突变,导致MDR3 蛋白缺失或表达降低。MDR3蛋白主要位于肝细胞毛细胆管膜上,为磷脂输出泵[4],其功能缺失会造成胆汁中磷脂缺乏,胆盐不能与磷脂构建混合微粒,游离的胆盐会对毛细胆管膜发生毒性去垢作用,从而导致胆管细胞受损,出现胆汁淤积、小胆管增生、炎症浸润,逐渐进展为门管区纤维化、肝硬化及门静脉高压,最后发展为终末期肝病。目前研究[5]表明ABCB4基因突变的类型与淤胆的严重程度相关:纯合的无义突变引起严重的PFIC-3;而杂合的无义突变、杂合的错义突变、纯合的错义突变则可能引起妊娠相关性肝内胆汁淤积或胆石症。本文2例经过基因检测证实在ABCB4基因处存在3处杂合突变点。家系验证分别来自其父母,兄弟俩有相似的临床症状,父母正常,推断在此家系中遵循隐性遗传规律,3处杂合突变为复合杂合突变,均为新发现的突变位点,软件预测结果提示有致病风险,诊断为PFIC-3。此家系中父母均为致病基因携带者,两个儿子均为复合杂合突变的概率仅为1/16,这个低概率事件的发生给家庭造成了极大的痛苦。
PFIC-3患者起病的年龄早晚不一,平均为3.5岁,瘙痒较轻微,其严重程度与黄疸程度不成正比,胆汁淤积呈慢性和进行性的特点,严重者可发生肝脾肿大、门静脉高压症、食管静脉曲张破裂出血,常死于肝衰竭,无肝外表现。婴儿多以黄疸、瘙痒、白陶土样便为首发症状,且常在儿童期就进展为肝硬化,而年龄相对较大的儿童常以肝脾肿大、胃肠道出血等肝硬化及门静脉高压表现为首发症状。本研究中2患儿均起病较早,进展快。哥哥4岁发病时病理显示活动性肝硬化,弟弟10月龄时病理提示早期肝硬化,有瘙痒症状,皮肤粗糙,全身可见散在抓痕,黄疸不明显,查转氨酶、TBA、GGT均偏高,符合PFIC-3的临床特点。PFIC-3肝穿刺病理并无特异性,可表现为纤维化、小胆管增生及炎症浸润、胆汁淤积、肝硬化,这些病变与其他淤胆性肝病不易区分。PFIC-3患者免疫组化可发现肝细胞毛细胆管膜上MDR3表达缺失[6],但并非病理常规检查内容。本文2例肝穿刺病理提示铜染色阳性,病理报告倾向肝豆状核变性,但实际上肝豆状核变性并不能靠病理表现确诊,肝铜定量才是有价值的诊断依据之一。有文献[7-8]报道证实某些胆汁淤积时间较长的PFIC-3患者可能有肝组织大量铜沉积和尿铜增加。
此外,PFIC-3因胆汁内缺少磷脂,易合并胆结石,脂溶性维生素及营养物质吸收不良,常可继发腹泻、生长发育迟缓、维生素K 缺乏性出血、维生素E缺乏性神经肌肉功能异常等[9]。本文2例患儿均未发生上述情况。研究[10-11]表明ABCB4/MDR3缺乏患者可发生肝胆恶性肿瘤。哥哥2013年1月腹部CT提示肝S6占位性病变,不除外肝癌,建议行活检病理以明确诊断或肝移植,但因家属经济困难拒绝行进一步检查及治疗,之后定期复查占位未见明显增大,仍随诊中。

表2 病例2历次生化检查结果
PFIC-3的治疗包括药物治疗:熊去氧胆酸(10~20 mg·kg-1·d-1)疗效相对确切,是本病最初的治疗策略,可改善肝功能,延缓肝硬化的进展,推迟肝移植时间,而苯巴比妥、考来烯胺、利福平等可改善瘙痒症状,研究[12]表明小分子ASBT抑制剂SC-425和A4250能有效降低Mdr2敲除小鼠(PFIC-3 动物模型)血清TBil水平,改善肝纤维化和炎症反应,其中A4250已成功通过了Ⅱ期临床试验,目前正在进行Ⅲ期临床试验;适当补充脂溶性维生素A、D、E、K、中链脂肪酸及钙等可以满足患者生长发育所需;胆汁分流手术多数可改善肝功能,肝移植是治疗PFIC-3最有效的方法。大部分PFIC-3患者会发展至肝病晚期,10年内需要行肝移植[7,13]。本研究中病例1于11岁得到确诊,之后虽经药物治疗但病情仍持续进展,14岁已发展至肝硬化晚期,肝脾大,脾功能亢进明显,门静脉高压伴侧支循环开放,肝占位性病变;而病例2发现、诊断早(2岁时),经熊去氧胆酸等治疗后肝硬化并无明显进展。因此PFIC-3早期诊断及干预十分重要,尽早予熊去氧胆酸等干预治疗可能会延缓疾病进展。此外随着PFIC基因突变谱的扩展以及常见突变对蛋白表达或功能损害的研究进展,根据患者基因型的特异性进行个体化治疗将成为趋势,如研究[14]表明使用杂交重组腺相关病毒-piggyBac基因疗法可改善胆汁淤积并降低PFIC-3型小鼠模型的致瘤性,此外腺相关病毒介导的基因疗法可使无胆汁酸饮食下的PFIC-3小鼠模型不会出现PFIC-3症状[15]。
在本研究中得到了一些经验教训,病例1在疾病起始阶段属于未明原因的胆汁淤积性肝病,肝穿刺病理提示铜染色阳性,病理报告倾向肝豆状核变性,化验24 h尿铜偏高,外院行肝豆基因学检测提示存在1个变异位点杂合子,初期拟诊为“肝豆状核变性”。直到弟弟又出现类似病情才促成了基因检测,发现了ABCB4的突变,最终明确诊断。家庭经济困难是基因检测推迟的原因,但医师对此检查的坚持力度也非常重要。所以临床上对于未明原因的GGT升高的胆汁淤积性肝病,需考虑PFIC-3可能,并及早建议患者做适当的基因分析,早日明确诊断,恰当治疗。
综上所述,本文报道的2例PFIC-3患者,黄疸不明显,但有轻度瘙痒、转氨酶升高、TBA明显偏高、GGT升高、小胆管增生等特征。新变异c.2570C>T(p.T857I)、c.2212A>T(p.I738F)和c.1694C>G(p.T565R)的发现为确诊和家系遗传咨询提供了遗传学依据。
