蛋白质二级结构预测服务器PSRSM
2020-07-09韩心怡刘毅慧
韩心怡,刘毅慧
(齐鲁工业大学(山东省科学院) 计算机科学与技术学院,济南 250300)
蛋白质二级结构[1]预测是生物信息学领域一项非常重要的研究课题,蛋白质二级结构不仅是构成蛋白质稳定构象的基础,同时也是进一步研究蛋白质三级结构的重要环节[2]。随着越来越多的蛋白质序列顺利完成了结构测试,国际上也不断有新的蛋白质二级结构预测方法被提出,同时也提供了多种在线预测服务器。试验选取了七种在线预测服务器:PSRSM、Spider3、SPOT-1D、RaptorX、MUFOLD,Psipred和Jpred4,并将它们的预测结果从Q3、Sov、边界识别率、内部识别率、转角C识别率,折叠E识别率和螺旋H识别率七个方面进行了对比评估。上述七种在线预测服务器,均采用了各自不同的预测方法:PSRSM采用基于数据划分和半随机子空间的预测方法[3];Spider3使用长短时记忆网络和双向递归神经网络的混合模型[4];SPOT-1D结合了残余卷积网络和双向递归神经网络[5];RaptorX使用了深度卷积神经场[6];MUFOLD采用了一种名为深度初始-内部-初始的网络[7];Jpred4通过JNet[8]算法提供预测,还有使用前馈神经网络的Psipred[9]。最新出现的PSRSM和SPOT-1D也增加了对大数据集的使用。
相比于文献[10],增加了对最新发布的SPOT-1D服务器介绍和评估,对所有服务器的使用流程做出了说明,同时增加了对转角C、折叠E和螺旋H、内部和边界结构的预测准确率评估,为研究者提供更多的参考角度。其中,各服务器Q3结果从高到低分别为PSRSM:89.96%;SPOT-1:88.18%;MUFOLD:86.74%;SPIDER3:85.77%;RaptorX:83.61%;Psipred:79.72%;Jpred4:78.29%。结果表明PSRSM预测效果优于其他服务器。
1 PSRSM-Server
PSRSM-Server是由齐鲁工业大学智能信息处理团队开发的蛋白质二级结构预测服务器,该服务器基于数据划分和半随机子空间(Partition and semi-random subspace, PSRSM)方法进行预测[3]。方法的主要流程为:首先根据蛋白质序列的长度将训练集划分为6种子集,然后用半随机子空间方法生成子空间,将SVM作为基本分类器,在子空间中训练基本分类器;最后通过多数投票规则把子集中的基本分类器结合,生成最终的分类器。网络输入为PSI-BLAST程序生成的20×L的PSSM矩阵,其中20为氨基酸个数,L为蛋白质长度。输入的蛋白质序列将会根据长度选择合适的分类器进行预测。此服务器将预测结果根据“H、G、I转为H”,“B、E转为E”,“其他结构转为C”的规则得出最终的3态结果。该方法在ASTRAL和CullPDB数据集上选取了15 696条去除较高相似度的数据上进行训练,在测试集CASP10、CASP11、CASP12、CB513,25PDB和T100(2018年2月前的100条)上Q3识别率分别达到85.51%、85.89%、85.55%、84.53%,86.38%和85.09%的良好性能[3]。PSRSM-Server 网址为:http://210.44.144.20:82/protein_PSRSM/default.aspx。
该网站提供了单条序列预测和批量序列预测的功能,点击“Sequence”,按照图1所示,输入邮箱,便可进行单条作业提交。所支持的蛋白质长度范围为10~800。
提交成功后网站会分配一个Job ID,使用者可根据此Job ID、序列或者预留邮箱在网站左侧“Predicted result”中根据不同的方式进行结果查询,如图2所示。
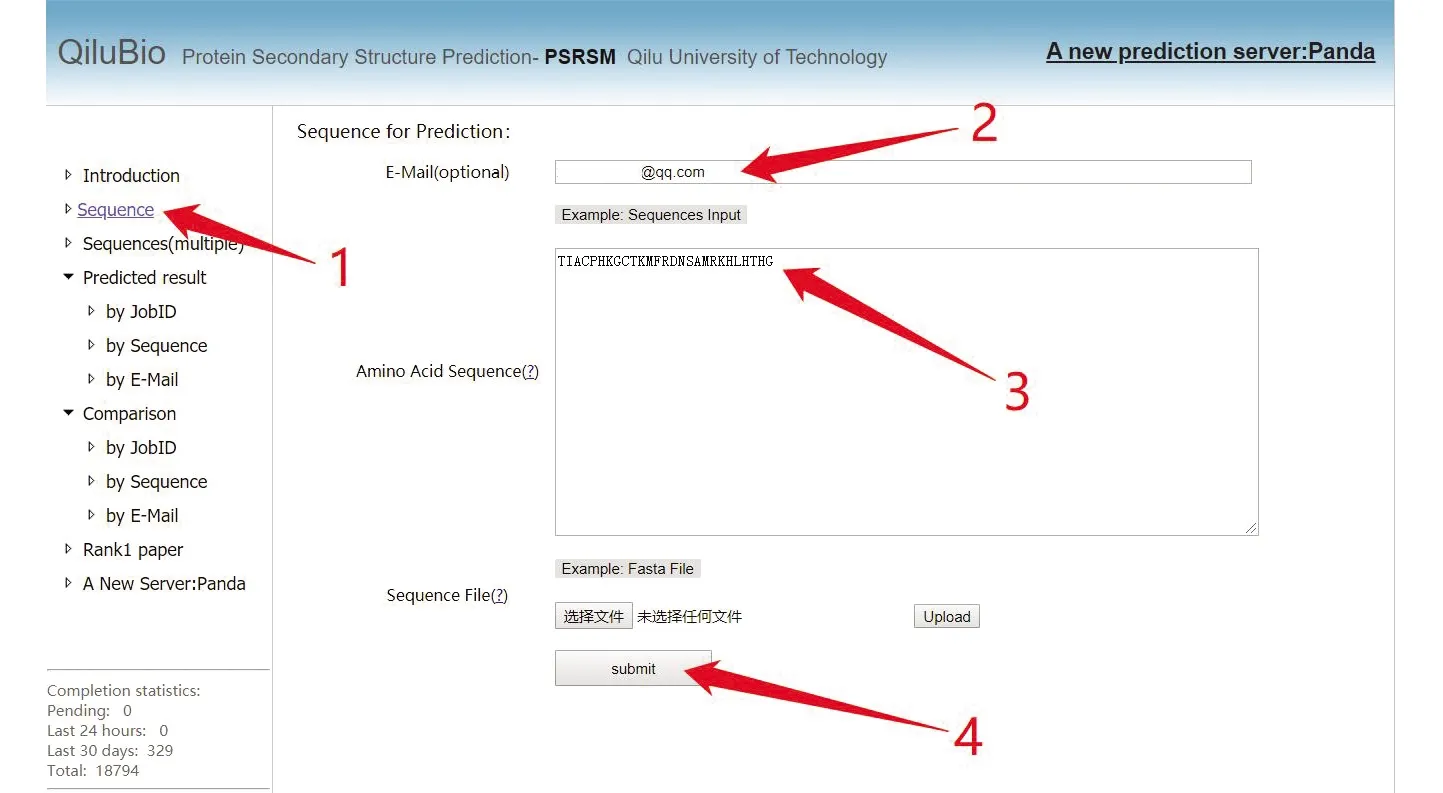
图 1 PSRSM 服务器单条数据测试提交步骤Fig.1 Single data test submission step of PSRSM-Server
注:用户可根据图中标注1点击“Sequence”然后输入查询邮箱,在标注3处输入提交序列,最后点击标注4处的“submit”完成单条数据提交.

图2 PSRSM服务器根据预留邮箱结果查询Fig.2 Query results based on email address in PSRSM-Server
注:用户首先在“Predicted result”处点击“by E-Mail”,然后在标注2处输入图1.2输入的预留邮箱,最后点击标注3处的 Search 即可查询测试状态.
用户可根据需要,选择点击“Download”下载结果或者点击“Select”在网页端查看结果,下载的结果将以txt格式保存。网站也同样支持上传Fasta格式文件进行预测,查询结果方式同图2。最后,该网站提供了查询预测准确率的功能,在左侧“Comparison”中,选中所需查询的结果,输入真实的DSSP,可直接查看Q3和Sov准确率。
2 其他方法介绍
2.1 Spider3
现有的机器学习方法在预测蛋白质二级结构时通常依赖于设置10到20个氨基酸残基大小的滑动窗口来捕捉“短到中”距离的残基相互作用,而该方法基于长短时记忆(Long short-term memory, LSTM)双向递归神经网络(Bidirectional recurrent neural network, BRNNs)[4],在不设置滑动窗口的情况下捕捉长距离的残基交互,改善了蛋白质二级结构的预测效果。该方法模型使用了两个节点数为256的双向递归神经网络层(BRNN),之后为两层节点分别为1 024和512的隐藏层。在BRNN层中采用了LSTM细胞来学习远距离和闭合序列内的依赖性。网络输入包括氨基酸的7种代表性理化性质(Physio-chemical properties, PP),PSI-BLAST的20维位置特异性评分矩阵(PSSM),以及来自HHBlits的30维隐马尔可夫模型特征。该方法数据集包含5 789个蛋白质,序列相似性截断值为25%,X射线分辨率低于2.0个Å。从所有数据中,随机选择4 590种蛋白质作为训练集(TR4590),其余1 199用作独立测试集(TS1199)。文献[4]中指出捕获序列的长距离相互作用可以使三态二级结构预测准确率达84%。
Spider3提供单条蛋白质和批量蛋白质序列预测的功能,同时网站也提供了预测软件下载的功能。在线提交测试序列过程中,由于服务器资源有限,同一个IP和邮箱下提交序列总数不可超过100条,注意提交序列过程中序列不要换行。Spider3网址为:http://sparks-lab.org/server/SPIDER3/。提交界面如图3所示。提交成功后,可在邮箱接收到最终结果,或者在网页端进行查看。
2.2 SPOT-1D
SPOT-1D是目前较新的一种蛋白质二级结构预测服务器。作为Spider3的改进方法,SPOT-1D在使用了双向递归神经网络的长短时记忆细胞(Long-Short-term memory Cells in Bidirectional recurrent neural networks, LSTM-BRNNs)基础上,结合了残余卷积网络(Residual Convolutional Networks, ResNets)[5],用来识别和传播整个序列中的短期和长期依赖关系,预测结果准确率得到了明显的提升,网络模型的描述在文献[5]的补充部分有详细的说明。该模型的特征输入由氨基酸的7种代表性理化性质,SPOT-Contact的预测接触图信息,PSSM和隐马尔科夫模型特征组成,共57维特征输入。相比于Spider3,SPOT-1D的预测更加准确,除了模型的改进,SPOT-1D从PISCES服务器中选取了更多数量的10 029条蛋白质进行训练。使用界面和操作方法同Spider3,但每次提交序列不可超过5条。SPOT-1D的网址为:http://sparks-lab.org/jack/server/SPOT-1D/。
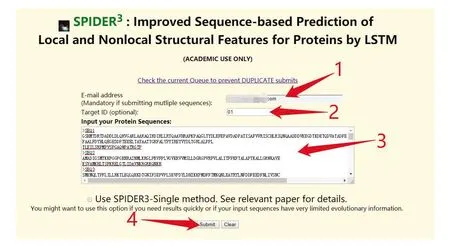
图3 Spider3提交测试序列Fig.3 Submission test sequence in Spider3
注:首先,用户在图中标注1处输入预留邮箱,在标注2处输入工作名称,然后在下方标注3处根据图中示例格式输入序列,最后点击标注4处的“Submit”完成序列提交.
2.3 RaptorX
RaptorX采用了名为深度卷积神经场(Deep convolutional neural fields, deepCNF)[6,11]的预测方法,该方法是深度卷积神经网络(Deep convolutional neural networks, DCNN)和条件神经场(Conditional neural fields, CNF)相结合。它能以分层的方式对复杂序列的结构关系进行建模,而且可以根据相邻残基之间的相关性建模。在DeepCNF中使用DCNN替换CNF中的浅层神经网络,以便捕获输入维度和输出标签之间的复杂关系,特别是对于在PDB中没有紧密同源性或具有稀疏序列谱的蛋白质具有很好的预测效果。针对紊乱蛋白质序列的预测,RaptorX在网络中增加了ROC曲线下面积最大化(Area under the ROC Curve , AUC)方法训练[12]。该网络的特征输入由21维PSSM和具有21个元素的二进制向量(表示第i个位置上的氨基酸)组成,共42维。RaptorX使用了CullPDB中5 600条蛋白质用作训练。该网站提供了批量预测的功能,提交方式如图4所示。在“My Jobs”里输入测试时提交的邮箱,等待结果链接。在线服务网址为: http://raptorx.uchicago.edu/StructurePrediction/predict/。
2.4 MUFOLD
MUFOLD采用名为Deep3I的网络(Deep inception-inside-inception networks, Deep3I)[7]进行蛋白质二级结构预测。Deep 3I由两个嵌套的可进行卷积操作的初始模块、卷积以及完全联通的致密层组成,有效地处理了氨基酸之间的局部和全局相互作用。MUFOLD对训练集输入特征有非常细致的设计,训练集为由氨基酸理化性质,PSI-BLAST特征和HHBlits特征组成的维度为58的特征向量。随机选取了CullPDB中的9 000条蛋白质用作训练集。该团队同时也利用初始胶囊网络的深度神经网络(Inception capsule networks)改善蛋白质γ-转角预测[13]。测试过程如图5所示:输入邮箱和项目名称后,在下方勾选 “Secondary Structure (3-states and 8-states)”,然后提交蛋白质序列,不允许序列字符断开或换行,且最多允许提交10个序列,每条序列的长度范围为30到700。该服务器网址为:http://mufold.org/mufold-ss-angle/。

图4 RaptorX批量提交测试序列Fig.4 Batch submission test sequence in RaptorX
注:首先,用户在图中标注1处输入预留邮箱,在标注2处输入工作名称,然后在下方标注3处根据图中示例格式输入序列,最后点击标注4处的“Submit”完成序列提交.

图5 MUFOLD提交测试序列Fig.5 Submission test sequence in MUFOLD
注:首先,用户在图中标注1处输入预留邮箱,在标注2处输入工作名称,然后在下方标注3处选择“Secondary Structure (3-states and 8-states)”,然后在标注4处的文本栏中输入提交序列,最后点击下方“Submit”完成序列提交.
2.5 Psipred
Psipred是常用的一种蛋白质二级结构预测服务器,该服务器聚合了多种蛋白质注释工具,提供分析方法作为软件下载。例如提供了序列和结构注释方法:Psipred,GenTHREADER,pGENTHREADER等。在网络结构方面,Psipred采用了两层前馈神经网络的体系,经交叉验证对网络性能进行评估。网络的输入是来自PSI-BLAST的20维特征矩阵。预测蛋白质二级结构的使用方法为:选择好所使用二级结构预测服务,然后输入序列,同样需要注意序列字符串不要换行,最后在输入的邮箱中接收结果。如图6所示。该服务网址为:http://bioinf.cs.ucl.ac.uk/psipred/。

图6 Psipred批量提交测试序列Fig.6 Batch submission test sequence in Psipred
注:首先,用户在图中标注1处选择服务“PSIPRED V3.3”,在标注2处的文本栏中输入提交序列, 在标注3和4处分别输入预留邮箱和工作名称,最后点击下方“predict”完成序列提交.
2.6 Jpred4
Jpred4通过JNet[8]算法提供预测。在上个版本JPred3[14]中使用JNet2.0对蛋白质序列进行预测,JNet2.0不使用频率文件,以PSSM和隐马尔科夫特征作为输入,使用两层来自SNNS神经网络包的人工神经网络,将隐藏层单元从9增加到100。Jpred4则基于JNet2.0神经网络的预测器进行了重新训练,通过使用1 358个SCOPe/ASTRAL v.2.04超家族域序列中的每个序列的一个代表进行7倍交叉验证来制作JNet2.3.1,通过搜索UniRef90v.2014_07生成PSI-BLAST构建了每个序列的多重比对。除了对JNet2.0重新训练之外,JNet中的HMM构建步骤已更新为HMMer3。Jpred4最终在150个未用于训练的超家族序列的盲测中评估其准确性,Q3准确率可达到82%。
该网站提供了批量预测的功能,如果只是提交单个序列则要在“Advanced options”中的“Select type of input”选项中,选中“Single Sequence”的“Raw/Fasta”模式;如果是批量在网页中输入蛋白质序列,则需要在“Single Sequence”下选中“Batch Mode”模式,然后输入接收结果的邮箱以及项目名(其中命名方式只可以是由字母数字和“_”字符组成)。批量提交过程如图7所示。最终结果将会发送到邮箱中,也可以在网页端等待查看。Jpred4提供服务的网址为:http://www.compbio.dundee.ac.uk/jpred4/index.html。

图7 Jpred4批量提交测试序列Fig.7 Batch submission test sequence in Jpred4
注:用户首先在标注 1 处输入需要提交的序列,如果同图中一样提交批量测试,则在标注2处选择“Single Sequence”下的“Batch Mode”模式;如果是单个序列提交,则选择“Raw/Fasta”模式,然后在标注3 和4处分别输入预留邮箱和工作名称,最后在标注5处点击“Make Prediction”完成工作提交.
3 结果评估
对上述七种服务器进行了预测结果评估,为保证实验数据量和公平性,测试集选取了PDB中2018年8、9、10、11月份发布的蛋白质[3,5,7],从中随机选取了60条30%同源性,60条40%同源性和60条70%同源性的蛋白质分别进行实验,最后又做出了这180条蛋白质的整体评估结果。实验数据集见表1。
评估采用了七种衡量标准,分别为Q3[2-14],Sov[2-6],边界识别率[3],内部识别率[3]和C、E、H每种独立结构识别率[4-5]的衡量标准。
3.1 Q3
在8态DSSP[15]中,根据 “G、H、I转为H(螺旋)”,“B、E 转为E(折叠)”,“其他结构转为C(转角)”将8态转为3态结构。Q3为正确预测的氨基酸数占所有氨基酸的比例,计算公式如下:
其中,QC为正确预测的转角数,QE为正确预测的折叠数,QH为正确预测的螺旋数,S为总的氨基酸数。
3.2 Sov
Sov是一种基于重叠片段比值的度量方式,设观测到的所有结构片段标记为Sab,所有预测到的片段则标记为Spr,而Sa是Sab和Spr状态相同的片段。任何观测到的残基长度被定义为length(Sab),对于Sa中任意一对片段,实际长度为minov(Sab,Spr),至少有一个残基的长度总限度为maxov(Sab,Spr)。基于以上定义,Sov的计算公式如下:
其中增设因子σ(Sab,Spr),允许蛋白质结构中的观测片段边界处的变化,其定义为:


表 1 180条数据集Table 1 180 data set
3.3 边界识别率和内部识别率
假设在一条长度为N的蛋白质序列中,第n(1 根据蛋白质同源性分类的所有服务器Q3、Sov、边界准确率和内部准确率见表2~表4,180条蛋白质的各项预测平均值见表5。 从表2可以看出,同源性30%的蛋白质数据集中,PSRSM在Q3、边界识别率和内部识别率上取得了最好的结果,分别达到了89.49%,84.25%和90.91%,并且对转角C和折叠E的识别率也是最好的,准确率分别达到了87.19%和90.27%。而SPOT-1D在Sov和螺旋H的识别率上结果要比PSRSM好一些,分别为83.16%和91.36%。 表2 30%同源性数据集Table 2 30% homology data set % 表3里40%同源性的数据下,PSRSM各项指标均为最好的结果,分别为Q3:90.53%;Sov:84.71%;边界识别率:85.24%;内部识别率:91.25%;转角C:87.34%;折叠E:88.46%;螺旋H:92.91%。SPOT-1D紧随其后,Q3为88.52%,相差2.01%,但Sov的表现依旧很出色,比PSRSM低约0.5%。 在表4中,对于70%同源性的蛋白质,PSRSM除了内部识别率,其他指标均取得了最好的结果,分别为:Q3:89.87%;Sov:86.12%;边界识别率:83.65%;转角C:89.08%,折叠E:88.34%和螺旋H:89.64%。SPOT-1D的内部识别率为91.46%,其他指标同PSRSM的差距和在40%同源性数据集的结果没有太大差别,约低1%~2%。 表5为全部数据集的评估结果,PSRSM各项指标全部取得了最好的结果:Q3:89.96%;Sov:84.52%;边界识别率:84.37%;内部识别率:91.18%;转角C:87.88%,折叠E:88.98%和螺旋H:91.25%。 表3 40%同源性数据集Table 3 40% homology data set % 表 4 70%同源性数据集Table 4 70% homology data set % 表 5 180条数据集Table 5 180 data set % 为了更加直观的对评估结果进行观察,将所有网站的Q3结果根据蛋白质长度做出了散点图,所选180条数据集中,蛋白质的长度范围为34-552,如图8所示。可以看出PSRSM(黄色)相对于其他颜色的位置更偏向于顶部,大部分服务器的预测准确率在70%~90%,PSRSM结果是优于其他服务器的。 表6对各服务器的预测方法、训练集、模型输入特征,Q3准确率和使用效率方面做了总结。本此测试从PDB中随机选取了一条长度为235的蛋白质:5XNE_A,测试各服务器从提交序列到获得结果的时间,结果为Jpred4最快,用时51 s;SPOT-1D所需时间最长,为16 m 42 s。然后又随机选取了五条蛋白质:5WOV_A(长度34)、5YIO_A(长度121)、5YKU_A(长度125)、5YVK_A(长度225)、5Y5B_A(长度228)做进一步的测试,结果为Jpred4用时最短,为2 m 25 s,SPOT-1D用时最长,为27 m 45 s。在WEB使用体验上,PSRSM、Spider3、RaptorX、Jpred4均提供了批量测试的功能;除了PSRSM,其他方法也提供了支持不同操作系统环境的软件下载服务。 图8 所有服务器的Q3散点图Fig.8 Q3 scatter plot for all servers 表 6 各服务器方法总结 ServersPSRSMSpider3SPOT-1DRaptorXMUFOLDPsipredJpred4方法原理分段特征提取+SVMsLSTM+BRNNsResNets+LSTM-BRNNsDCNN+CNFCNN前馈神经网络SNNS神经网络包训练集数量(ASTRAL+CullPDB)15 696(PISCES服务器中选取)4 590(PISCES服务器中选取)10029(CullPDB)5 600(CullPDB)9 000———(SCOPe/ASTRAL)1 358特征输入PSSM氨基酸7种理化性质+PSSM+隐马尔可夫模型特征氨基酸7种理化性质+SPOT-Contact预测接触图信息+PSSM+隐马尔可夫模型特征PSSM+21个元素的二进制向量氨基酸理化性质+PSSM+隐马尔可夫模型特征PSSMPSSM+隐马尔可夫模型特征Q3/%89.9685.7788.1883.6186.7479.7278.29是否支持批量预测是是是是否否是一条蛋白质(长度235)运算时间5 m 39 s3 m 40 s16 m 42 s3 m 19 s2 m 30 s1 m 46 s51 s五条蛋白质运算时间17 m 7 s12 m 39 s27 m 45 s7 m 5 s9 m 20 s6 m 30 s2 m 25 s 对PSRSM、Spider3、SPOT-1D、RaptorX、MUFOLD,Psipred和Jpred4七种在线服务的蛋白质二级结构预测效果进行了评估。整体来看,在多种比对方法下,PSRSM绝大多数指标都取得了最优的结果。从方法选择角度来看,PSRSM根据蛋白质长度划分不同子集和基于大数据集的训练方式,明显有较好的成效,而紧随其后的SPOT-1D多种深度学习方法和大数据集的训练结合,Sov的准确率也是非常稳定,效果出色。可以看出,蛋白质二级结构预测可以从结合多种深度学习方法,运用大数据进行模型训练做进一步的研究。




Table 6 Summary of methods for each server
4 结 论
