基于深度森林和DNA甲基化的癌症分类研究
2020-07-06郑一超侯维岩
刘 超,吴 申,郑一超,侯维岩
郑州大学 信息工程学院,郑州 450001
1 引言
近年来,癌症分类预测模型与生物学和遗传数据相结合,能够更准确地评估癌症风险[1]。DNA甲基化已经成为癌症研究中最重要的表观遗传修饰之一,研究表明,与“正常”组织相比,“肿瘤”组织中的DNA甲基化模式[2]异常。利用机器学习的理论和方法对癌基因相关的DNA 甲基化调控位点的识别,实现解析癌症的发生发展机制,识别新的癌症标记是一个生物信息领域的新研究方向[3]。
癌症基因组图谱(The Cancer Genome Atlas,TCGA)[4]是目前最全面的癌症测序数据库之一,其提供的丰富的癌症样本数据为开发癌症分类模型提供了前景。像大多数数据一样,TCGA中数据本质上是不平衡的。这些高度不平衡数据的分类受到多数类的影响,导致假阴性率增加[5]。
针对不平衡数据集癌症分类模型的上述问题,本文提出了一种混合采样的集成分类模型,利用SMOTE 算法扩充少数类样本集,通过Tomek Link 算法剔除边界数据和噪声数据,得到相对平衡的训练数据,将训练数据导入gcForest模型,在保证对于多数类的分类精度的前提下,有效地提高了对于癌症少数类样本的分类精度。
2 方法
本文所提出的基于混合采样的集成分类模型如图1所示,主要分为3个阶段:数据预处理、特征选择以及模型训练和验证。在预处理阶段使用SMOTE算法作为维持平衡类分布的方法,Tomek Link 欠采样算法用于数据清理,以消除噪音。为了减少数据的特征空间,仅考虑那些与癌症有因果关系突变的基因。数据通过COSMIC[6]和CIViC[7]在线数据库资源获得。使用gcForest 算法构建分类模型。该模型应用于6 种不同癌症类型。DNA甲基化数据来自https://portal.gdc.cancer.gov/repository。
2.1 数据预处理
2.1.1数据处理
TCGA 项目公布了28 种癌症类型的DNA 甲基化数据。原始数据(0 ≤x≤1)可通过TCGA 官网在线获取,并映射到特定的数据位置或范围(例如,chr19:19033575 指示染色体19 上的位置19033575)。本文使用了由 Broad Institute 的 FireBrowse 对 DNA 甲基化数据进行预处理,FireBrowse[8]将数值映射到基于HGNC命名法注释的特定人类基因[9]。每个样本文件用TCGA标识符值[10]注释,该值指示样本是肿瘤组织还是正常组织(例如,TCGA-2F-A9KW-01:肿瘤类型:01~09(1类),正常类型:10~19(0类)。本文研究中选取TCGA数据库中样本数据相对较大的6种肿瘤类型统计数据见表1。
2.1.2采样
表1表明从TCGA中获取的数据严重不平衡,这是由于目标类的分布不统一所致。目前使用的分类方法对于癌症样本实现非常高的准确性,但对正常样本的敏感性较低[11]。因此,本文提出了一种混合采样模型,最大限度地提高对正常样本的敏感性,同时实现较高的准确性。

表1 本项目中使用的DNA甲基化数据
(1)合成少数类采样技术(SMOTE)
SMOTE(Synthetic Minority Oversampling Technique)由Chawla[12]提出,是基于随机过采样算法的一种改进方案,主要思想是将新样本插入少量相似样本中以平衡数据集。 SMOTE 算法不是简单地复制样本的随机过采样方法,而是添加了一个不存在的新样本,因此在某种程度上可以避免过多的分类过滤。SMOTE算法的基本原理如下所示:
①对少数类中每一个样本x,以欧氏距离为标准计算它到少数类样本集中所有样本的距离,得到其k近邻。
②根据样本不平衡比例设置一个采样比例以确定采样倍率N,对于每一个少数类样本x,从其k近邻中随机选择若干个样本。
③对每一个随机选出的近邻,与原样本按公式(1)构建新的样本。
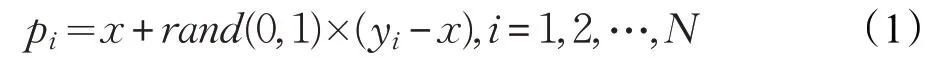
其中,x是样本,rand(0,1)表示区间(0,1)内的随机数,yi是k个最近邻。
(2)Tomek Link
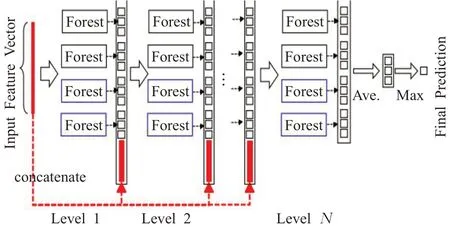
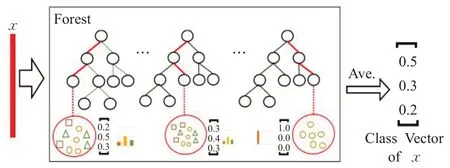
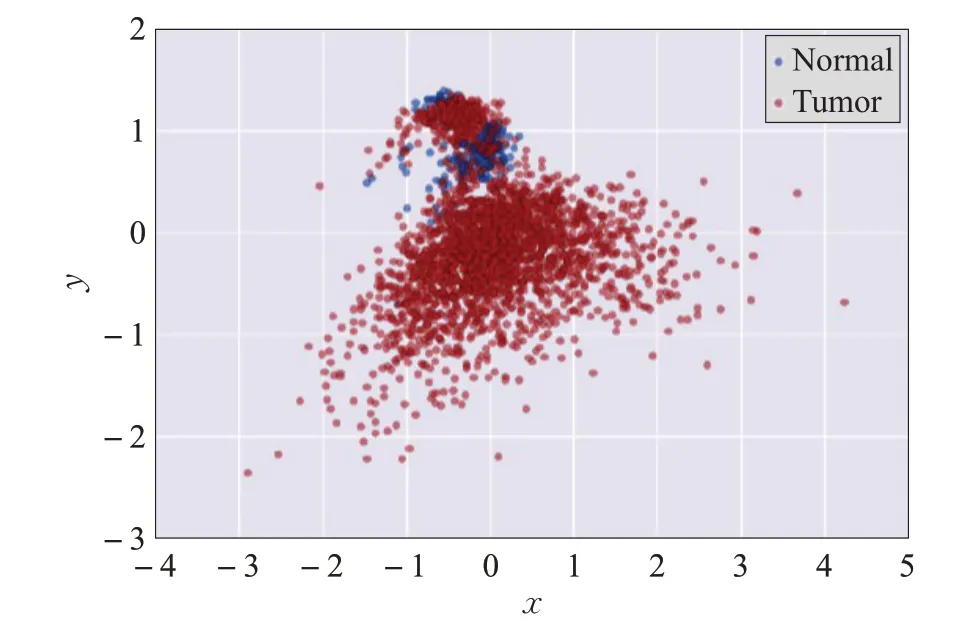
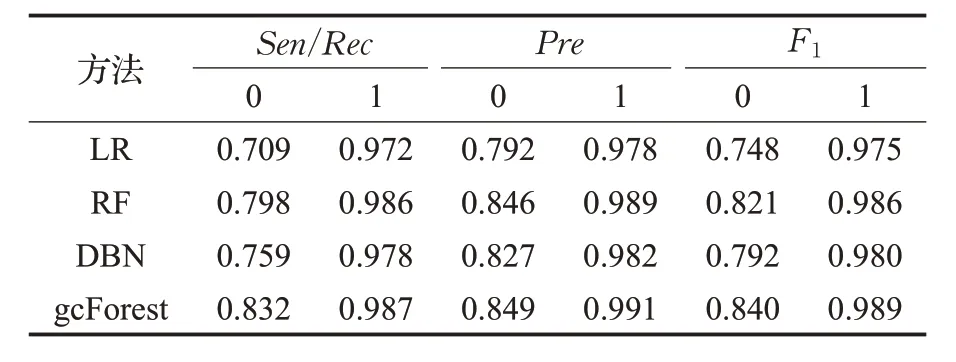
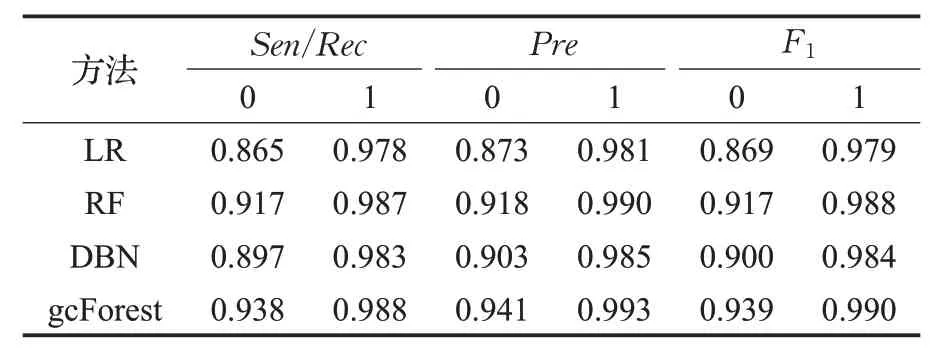
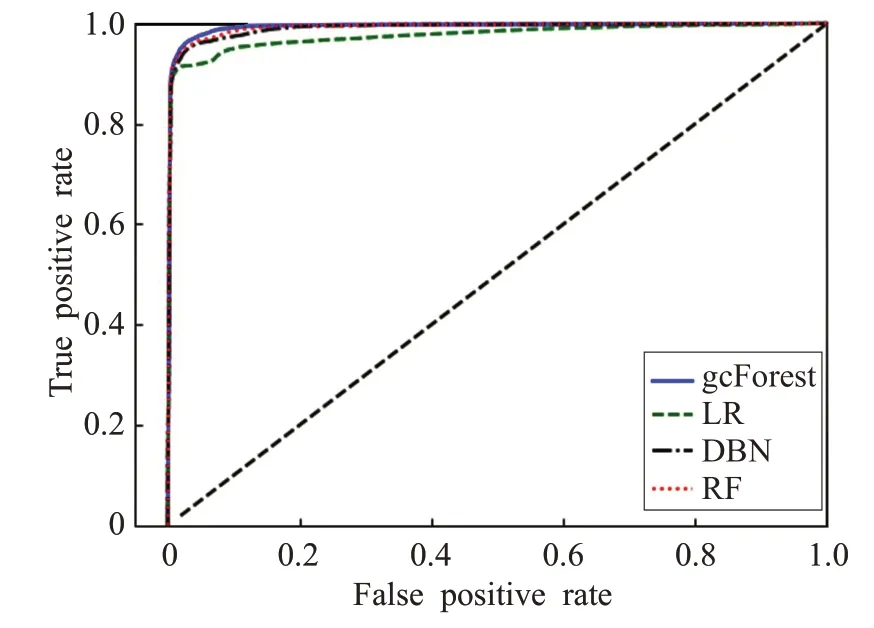
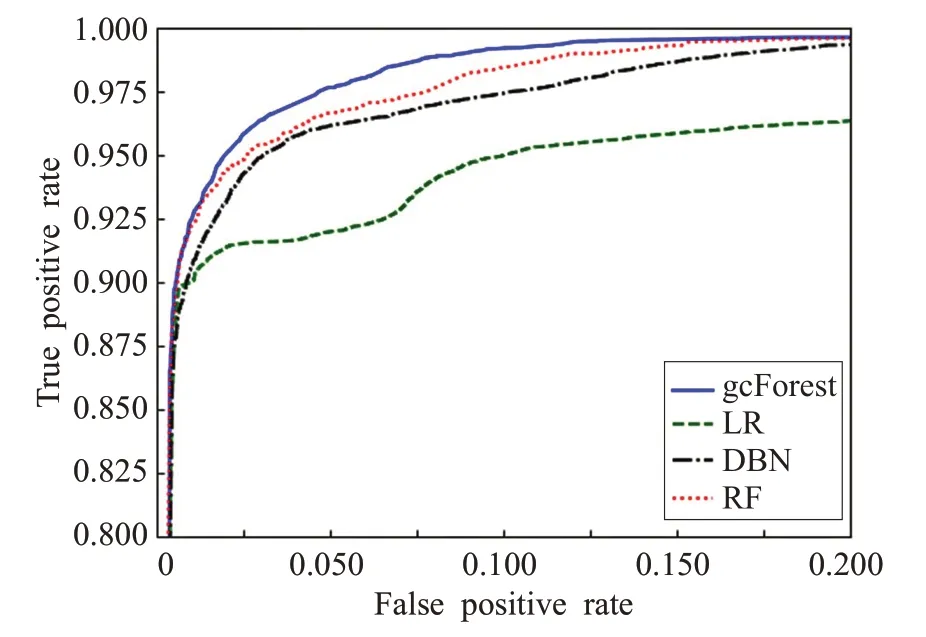
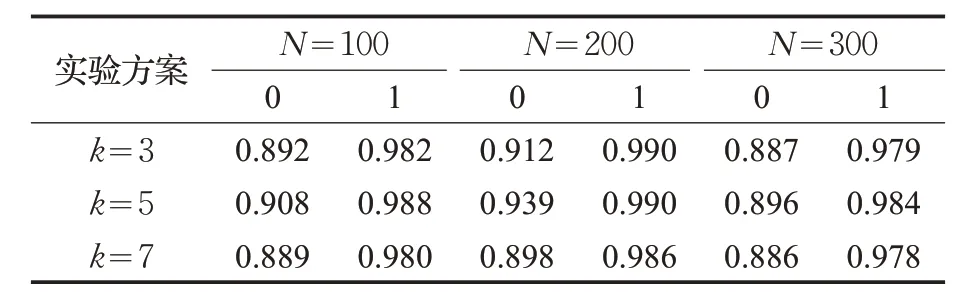
由于SMOTE方法在平衡类别分布的同时也扩张了少数类的样本空间,产生的问题是可能原本属于多数类样本的空间被少数类“入侵”,容易造成模型过拟合。为解决此问题,采用Tomek Link 算法[13]剔除噪声点或者边界点,可以很好地解决“入侵”的问题。Tomek Link算法的核心思想是:假设样本点xi和xj属于不同的类别,d(xi,xj)表示两个样本点之间的距离。称(xi,xj)为一个Tomek Link对,如果不存在第三个样本点xl使得d(xl,xi) 图1 本文研究的技术流程图 此外,本文研究是通过插入样本点与原样本点和其近邻之间的欧式距离来保证插入数据与原样本具有较好的相似性,在通过SMOTE算法对少数类样本扩充后,通过Tomek Link 算法对其欧式距离进行判断,剔除那些相似性低的样本点,即文中称为噪声点或者边界点。 TCGA中各种癌症类型包含的DNA甲基化数据有大于20 000 个蛋白质编码基因作为其特征变量。在这种情况下,特征选择是非常重要的[14]。因此,该项目只针对那些已被生物学鉴定为具有癌症突变意义的基因。这些基因是通过癌症基因普查(The Cancer Gene Census,COSMIC)和癌症变异的临床解释(Clinical Interpretation of Variants in Cancer,CIVic)获得的。 gcForest算法是由南京大学周志华教授提出的一种由决策树组成的集成算法[15]。gcForest的核心主要包括两大块:级联森林(Cascade Forest)和多粒度扫描(Multi-Grained Scanning)。级联森林的构成:级联森林的每一层都是由好多个森林(既有随机森林,又有完全随机森林)组成,而每一个森林又是由好多个决策树组成。其中每一层的随机森林和完全随机森林保证了模型的多样性,具体的级联森林结构如图2所示。 图2 级联森林结构图 图2 中的级联森林每一层包括两个完全随机森林(黑色)和两个随机森林(蓝色)。每个完全随机森林包含30 个完全随机的决策树,每个决策树的每个节点都是随机选择一个特征做分裂,直至每一个叶节点包含的实例属于同一个类;每个随机森林也是30个决策树,每个决策树的生成是随机选择sqrt(d)个特征(d输入的总特征),每次选择基值最佳的做分裂,级联森林迭代到效果不能提升就停止。 每个森林中都包括好多棵决策树,每个决策树都会决策出一个类向量结果(以三类为例,下同),然后综合所有的决策树结果,再取均值,生成每个森林的最终决策结果是一个三维类向量,每个森林的决策过程如图3所示。这样,每个森林都会决策出一个三维类向量,回到图2 中,级联森林中的4 个森林就都可以决策出一个三维类向量,然后对4 个三维类向量取均值,最后取最大值对应的类别,作为最后的分类结果。 图3 每个森林的决策过程 召回率/敏感性——Sen/Rec的值越大,说明有病的被判为有病的越大,漏检(FN)越小。 精确率-查准率,即正确预测为正的占全部预测为正的比例。 F1为算术平均数与几何平均数的比值,越大越好。 ROC 曲线是反应敏感性和特异性连续变量的综合指标,提供不同实验之间在共同标尺下的直观比较,ROC曲线越凸、越接近左上角表明其诊断价值越大,利于不同指标间的比较,曲线下面积可评价诊断准确性。 从TCGA 获取的DNA 甲基化数据按照训练集:测试集比例为7∶3。图4 显示了训练数据的PCA 二维图,从图中可以发现样本数据分布严重不平衡。 图4 采样前的分布 表2为gcForest、Logistic Regression(LR)[16]、随机森林(RF)[17]和深度置信网络(DBN)[18]4种分类方法的模型性能对比。由表2可以看出对于多数类样本4种分类算法都具有较高的准确性,但对少数类的敏感性较差,这是由于数据内部的不平衡造成的。 表2 混合采样前的4种模型性能指标 为解决上述问题,本文提出的SMOTE 算法结合TomekLink 算法的混合采样模型对DNA 甲基化数据进行预处理,处理后DNA 甲基化数据的PCA 二维图如图5所示,数据分布相对平衡。 图5 采样后的分布 数据标准化后,再次对比4 种分类模型,如表3 所示,使用本文提出的混合采样模型后,4种分类模型对于少数类的评价指标Sen/Rec、Pre和F1都得到较大程度的提升。 表3 混合采样后的4种模型的性能指标 对比表2和表3还可以发现,4种分类模型中,无论是采样前还是采样后,gcForest 算法的分类效果最好,为了清晰直观地对比4 种分类模型的性能,如图6 和图7 所示,为4 种分类模型的ROC 曲线,对比表明深度森林gcForest 算法的性能最佳。这是由于本文研究使用的DNA 甲基化测序数据维度高,gcForest 算法中的多粒度扫描结构通过采用滑动窗口对输入数据特征进行预处理,其表征学习能力得到进一步的提升。其次,将得到的特征输入到gcForest 算法的级联森林中进行训练,级联森林将输入特征与原始特征结合,通过两层级联森林中的随机森林和完全随机森林的学习,相比于逻辑回归、随机森林和深度置信网络而言,能够更加充分地学习特征之间的相关性,因此获得的性能最佳。此外,相比于深度置信网络,gcForest 算法的模型参数更少,容易训练,其在癌症分类研究中的小数据集方面更具优势。 图6 4种分类模型的ROC曲线 图7 ROC曲线图左上方详细图 此外,本文研究中还针对不同近邻k和采样倍率N在gcFores 分类模型中对综合评价指标F1的影响进行了对比分析如表4所示,在9组参数设置组合中,N=200(%),k=5 时的性能最佳。 表4 不同近邻k 和采样倍率N 对F1 的影响 分析其原因主要有两点:(1)采样倍率N=100 时,平衡化后的正、负样本数据依然具有较大的不平衡性,使得实验结果改善不明显。当采样倍率N=300 时,平衡化后扩充的样本数远大于原始样本,由于SMOTE 算法等各种过采样操作实质上是“无中生有”,平衡化后对模型的性能反而不明显。(2)关于最近邻k的选择,当k=3 时,模型复杂度高,容易产生过拟合现象,学习的估计误差增大;当k=7 时,虽然降低了学习误差,但由于样本数据集小,k取7时与样本较远的数据也会对模型的分类结果产生作用,增大了模型学习的近似误差。 本文提出了一种基于混合采样的不平衡数据集成分类模型,该模型使用过采样SMOTE算法和TomekLink算法混合采样,并结合深度森林gcForest算法构建了癌症分类模型。通过对比Logistic Regression、随机森林和深度置信网络DBN等模型,实验结果表明,本文提出的基于混合采样的不平衡数据集成分类模型在保证对于多数类的分类精度的前提下,有效地提高了对少数类的敏感性。此外,gcForest 分类模型在小规模不平衡数据集中的应用较之深度置信网络DBN等性能更佳。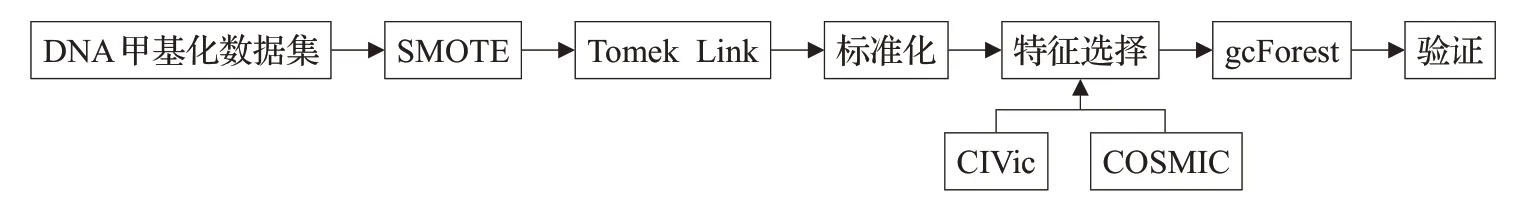
2.2 数据预处理
2.3 分类模型
2.4 评价指标

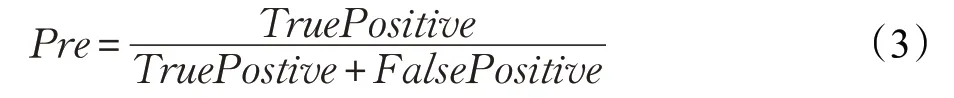

3 分析与讨论

4 结论
