磷酸化修饰位点预测分析研究
2020-07-06邓晓政徐瑞杰陈宇
邓晓政,徐瑞杰,陈宇
(青岛大学 数据科学与软件工程学院,山东青岛 266071)
蛋白质磷酸化修饰是生物体内一种普遍存在的翻译后修饰类型[1],至少有三分之一的细胞蛋白中存在磷酸化修饰[2]。该过程具有可逆性,与许多细胞内生化过程相关,如蛋白-蛋白相互作用[3]、免疫反应[4]、有丝分裂/细胞周期[5]等。磷酸化修饰的失调可能会导致多种疾病发生[6]。研究发现,大多癌症以及肌肉疾病与磷酸化位点及其协同位点的异常修饰相关[7]。因此,准确定位磷酸化修饰位点对人类疾病的研究具有重大意义。
近年来,磷酸化修饰位点的定位更多地依赖于机器学习算法,如支持向量机、人工神经网络、隐形马尔可夫模型和随机森林等。2014年,Dou等人[8]通过8种不同的序列特征打分以及支持向量机算法构建预测模型。Hamid等人[9]在2016年开发了一种基于序列结构属性以及随机森林算法的磷酸化位点预测工具。2017年,Tan等人[10]以Dou的磷酸化位点数据为模板开发了基于位置的卡方表特征和伪位置特定评分矩阵的支持向量机分类器。虽然结合机器学习算法对磷酸化修饰位点的预测取得了一定进展,但是许多模型不具备跨物种普适预测能力,对不同物种的预测存在偏差。因此针对于单个物种利用机器学习算法开发精度更高的蛋白质翻译后修饰位点预测器十分必要。
本文提出了一种专门用于定位人类磷酸化修饰位点的预测方法,该方法采用动态筛选机制针对数据集选取最优序列氨基酸理化性质和氨基酸组成等特征,并基于支持向量机算法进行磷酸化修饰位点的准确定位,相关算法以及预测工具命名为HPSP(Human Phosphorylation Site Predictor)。
1 材料与方法
1.1 材料
从UniprotKB/Swiss-Prot中下载经过验证的人类蛋白质序列数据共计20 402条,筛选出关于丝氨酸(S)、苏氨酸(T)和酪氨酸(Y)磷酸化位点的蛋白质序列。使用CD-HIT程序去除同一性阈值为30%的冗余序列,最后获得丝氨酸、苏氨酸、酪氨酸各4 917条、2 002条以及609条蛋白质序列片段。
分别以磷酸化位点为中心,截取前后各13个氨基酸残基的对称窗口作为编码特征的磷酸化序列片段,窗口内残基总数为27个。如果磷酸化位点在N端或C端附近,使用大写字母“O”代表缺失的氨基酸。经过上述处理后分别得到3种序列片段各22 763条、3 805条和1 122条作为正样本集。假设在同一蛋白质上没有任何磷酸化信息标记的丝氨酸/苏氨酸/酪氨酸残基都是非磷酸化位点,本文将同一蛋白质中没有任何磷酸化信息标记的丝氨酸/苏氨酸/酪氨酸残基作为负样本集。从各自蛋白质序列片段中截取未被磷酸化的丝氨酸、苏氨酸、酪氨酸前后各13个残基的对称窗口作为负样本集,分别为319 176条、91 936 条、11 628 条。
为避免正负样本集数目极不平衡导致模型过拟合的情况,对负样本集进行随机抽样,使正负样本集保持相同的数量规模。
1.2 方法
1.2.1 物理化学属性特征
氨基酸的物理化学属性(PCP)如疏水性、分子量、可及表面等,在蛋白质的结构和功能研究中具有重要作用。蛋白质序列的结构和功能在某种程度上与组成蛋白质的每一个氨基酸的物理化学属性都有很大关联。而且PCP已经成功应用于蛋白质磷酸化修饰位点预测工作中[11]。从AAindex数据库[12]中获取544种物理化学性质,利用F值检验进行物理化学属性的筛选,具体公式如式1所示。

式中,μi+、μi-分别表示正样本和负样本中第i个物理化学属性的平均值,σi+,σi-分别表示正样本和负样本中第i个物理化学属性的方差。
某一个物理化学属性的F值越高,则认为该物理化学属性特征能很好地区分磷酸化位点和非磷酸化位点。对544个物理化学属性分别计算F值,取F值最大的前20个物理化学属性作为特征。由于AAindex中氨基酸指数范围较广,可能对训练模型的预测造成影响,因此对氨基酸指数进行归一化处理,具体的处理过程如式2所示。

式中,Pi表示某一物理化学属性在AAindex中的值,Pinorm表示经过归一化处理后的结果。Pmax、Pmin分别表示某一物理化学属性在AAindex中的最大值和最小值。
最后每一个序列片段可以得到20维物理化学属性特征 PCP(P)=(p1,p2,p3…p20)。
1.2.2 KNN距离
KNN(K最邻近分类算法)距离从可能的磷酸化位点周围局部序列中提取序列相似性信息,这些序列相似性信息反映了蛋白质序列结构上的差异。序列相似性越高,序列之间存在的功能就越相似。利用KNN距离提取相似性信息,首先计算未知蛋白片段到正负样本集的距离。距离D(S1,S2)定义为式3。

式中,p表示序列片段中心两侧氨基酸残基的数目,i表示序列片段中氨基酸的位置。Blo是基于BLOSUM62矩阵[13]得到的氨基酸相似性打分矩阵。具体的转换过程定义为式4。

其中,M表示BLOSUM62矩阵,min和max分别为BLOSUM62矩阵中的最小值和最大值。计算未知蛋白片段到正负样本集的KNN距离并排序后,选择前k个样本并统计这k个样本中正样本所占的百分比,即为最终的KNN特征值。KNN特征提取中,k值的选择对于分类效果具有很大影响,k值过大会使算法时间复杂度越高,而k值过小则可能丢弃真正有意义的序列。因此本文选取5个不同的k值,每条蛋白质序列可得到 5 维 KNN 特征 KNN(P)=(k1,k2,k3,k4,k5)。
1.2.3 信息熵与熵密度
1948年,Shannon[14]提出信息熵以度量给定系统信息含量。越是混乱的系统,信息熵越高;而越是有序的系统,信息熵越低。对于蛋白质序列片段而言,保守信息随着位置变化而变化,而磷酸化修饰位点附近的某些残基对磷酸化位点的识别有重要影响。因此信息熵是对蛋白质序列片段中各个位置残基保守性信息量的度量。将信息熵与熵密度作为衡量磷酸化可能性高低的标准,具体计算方法如式5和式6所示。

其中,X表示蛋白质序列片段,fi(X)表示在该蛋白质片段中第i个氨基酸残基的出现频率。最后每一条蛋白质序列片段得到1维的信息熵H(P)=x和20维的熵密度特征S(P)=(s1,s2,s2…s20)。
1.2.4 氨基酸组成
氨基酸组成(AAC)是最经典的蛋白质特征编码方法之一。Lee等[15]利用修饰位点周围的氨基酸组成信息作为预测蛋白质翻译后修饰位点的一个重要特征。AAC与每一个氨基酸的物理化学属性能够反映蛋白质序列的生物化学环境,与磷酸化序列片段具有相似的生物化学环境的片段很有可能存在潜在的磷酸化位点。每个蛋白质片段可被编码为一个20维的向量 AAC(P)=(a1,a2,a3…a20)。
2 结果与分析
本文使用LIBSVM进行预测模型的搭建。首先从已获得数据集合中选取部分数据来进行训练,分别从训练集中随机抽取十次正负样本,选取比例为1∶ 1。
在此基础上,对支持向量机径向基(RBF)核函数和c、g参数进行优化。在利用LIBSVM进行预测之前,利用grid.py得到最优cost值和gamma值,使用交叉验证对比选取RBF核类型和C-SVC类型来创建模型。RBF核函数如式7所示。

采用十倍交叉验证法对预测方法的性能进行了评价。利用精度(Pr)、灵敏度(Sn)、特异性(Sp)、准确度(ACC)和马太相关系数(MCC)对该预测系统的性能进行评价。MCC是反映正负样本成功预测的综合指标,其值范围为-1~1,数值越趋近于1表示预测性能越好,数值越趋近于-1表示预测性能越差。该模型在训练集上的预测结果如表1所示。
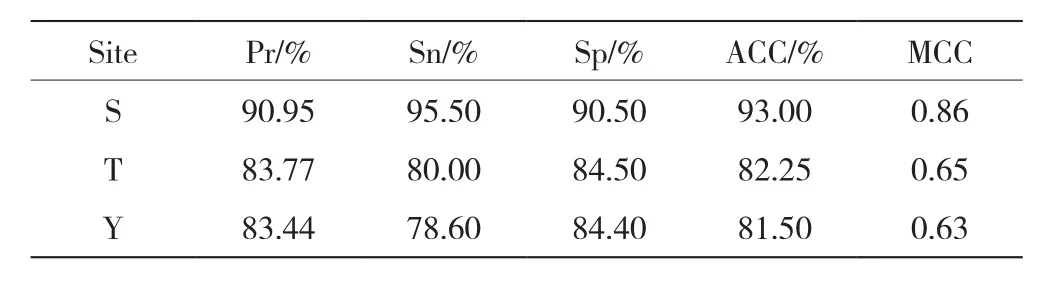
表1 训练集上的预测结果
3 讨论
3.1 特征分析
为了获得磷酸化修饰位点预测的最优特征,以酪氨酸正负样本集为例,进一步验证四类特征单独预测以及全融合特征预测在训练集上的性能,具体表现如表2所示。对于单个特征预测而言,KNN距离特征的预测性能最优,MCC为0.54,比其他三类特征预测的MCC值高0.2左右,这也印证了前述特征分析中正负样本KNN打分值差异性较大的结论。虽然全融合特征的Sp值相比于KNN距离特征没有明显的提高,但是在其他四个评价指标上存在显著提高。由此可见,四类特征全融合表现最优,最终选取四类特征全融合来搭建预测模型。

表2 基于不同特征的酪氨酸磷酸化修饰位点预测性能
3.2 工具对比
磷酸化位点预测工具有很多,为了进一步说明本算法在已知磷酸化位点预测上的优势,将本文提出的预测模型HPSP与RF-Phos方法在独立测试集上进行比较。从文献[16]中收集独立测试数据集,从400个磷酸化修饰位点中除去训练集中存在的数据,剩下的作为独立测试集。正样本数量分别为188条、82条、100条,负样本数量分别为364条、246条、128条。
为了更加客观地比较,使用四种评价指标对预测性能进行评估。在独立测试集上,虽然在酪氨酸上的MCC、ACC值与RF-Phos相当,但是在特异性Sp上有较大的提高。在丝氨酸与苏氨酸上,灵敏度和特异性都高于RF-Phos。三种磷酸化修饰的MCC与ACC值均高于RF-Phos,其中在苏氨酸磷酸化位点上的性能表现最优,准确率ACC提高了0.3%,MCC从0.56提高到0.63,是性能评估中一个全面的综合指标。表3所示的测试结果表明本模型HPSP整体优于RF-Phos的预测算法。
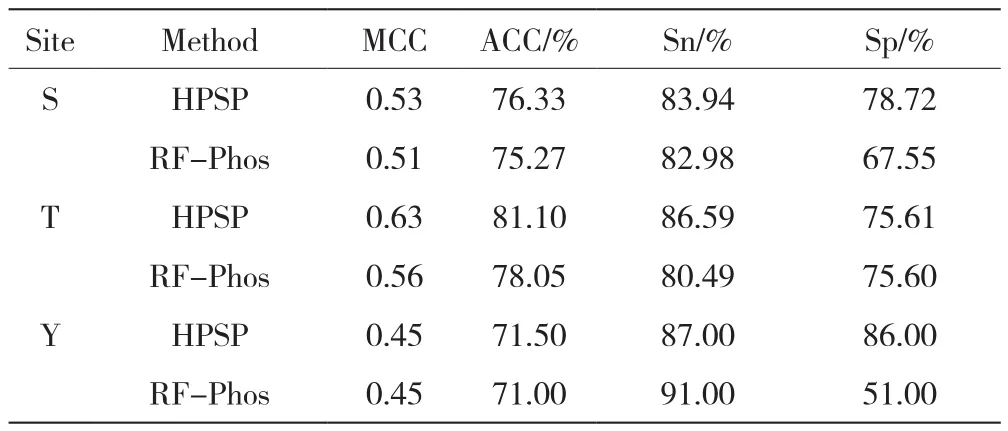
表3 模型HPSP与RF-Phos工具性能比较结果
该结果充分说明模型HPSP对磷酸化修饰位点的预测具有良好的性能。今后,可以探索其他的方法来进一步提高模型的预测能力,如增加其他具有代表意义的特征、改变滑动窗口长度大小或者配合其他机器学习分类预测算法等方法;同时也可以将激酶信息整合到预测模型中以识别激酶特异性磷酸化修饰位点。
4 结论
本文基于支持向量机开发了一种针对人类蛋白质序列数据的非特异性磷酸化位点预测方法HPSP,该方法整合了信息熵和密度熵等四类特征,利用F值检验方法筛选出能够显著区分磷酸化位点和非磷酸化位点的特征,利用支持向量机算法进行预测模型的搭建。在独立测试集,对丝氨酸、苏氨酸、酪氨酸磷酸化修饰位点的预测准确值分别达76.33%、81.10%、71.50%。
