Shared (epi)genomic background connecting neurodegenerative diseases and type 2 diabetes
2020-07-06ValerioCaputoAndreaTermineClaudiaStrafellaEmilianoGiardinaRaffaellaCascella
Valerio Caputo, Andrea Termine, Claudia Strafella, Emiliano Giardina, Raffaella Cascella
Valerio Caputo, Claudia Strafella, Emiliano Giardina, Raffaella Cascella, Department of Biomedicine and Prevention, Tor Vergata University, Rome 00133, Italy
Valerio Caputo, Andrea Termine, Claudia Strafella, Emiliano Giardina, Molecular Genetics Laboratory UILDM, Santa Lucia Foundation, Rome 00142, Italy
Andrea Termine, Experimental and Behavioral Neurophysiology Laboratory, Santa Lucia Foundation, Rome 00142, Italy
Raffaella Cascella, Department of Biomedical Sciences, Catholic University Our Lady of Good Counsel, Tirana 1000, Albania
Abstract
Key words: Type 2 diabetes; Alzheimer's disease; Parkinson's disease; Metabolism;Neurodegeneration; Neuroinflammation; Genetic variants; Epigenomic background;Disease interactome
INTRODUCTION
The recent progress in medicine and the improvement of health conditions have contributed to the rise of life expectancy, on the one hand. On the other hand, the better healthcare conditions and the availability of several therapeutic approaches have run in parallel to the progressive aging of populations, which raised novel challenges to be faced by the healthcare systems and the scientific communities. In fact, the progressive aging population has resulted in the increased prevalence of chronic pathologies, especially of metabolic, neurodegenerative and movement disorders[1]. In particular, type 2 diabetes (T2D), Alzheimer's disease (AD) and Parkinson's disease (PD) are among the most prevalent chronic, age-related pathologies that deserve particular attention given their dramatic impact on patient quality of life, their economic and social burden as well the etiopathogenetic mechanisms, which may overlap in some cases.
In fact, T2D accounts for 90% of cases of diabetes mellitus, which affects 285 million people worldwide[2]. It is mainly caused by a combination of insulin resistance and relative insulin deficiency[3], which results in glucose dyshomeostasis and other concomitant conditions, including hypertension and dyslipidemia[4]. AD is characterized by progressive loss of memory and cognitive domains responsible for functional independence[5,6]. This pathology accounts for the 60%-80% of overall forms of dementia and represents the sixth cause of death in the world. It affects about 30-46 million people[5,7-9], with an increasing prevalence depending on age (ranging from 0.3%-0.5% at age 60 to 11%-15% at age 80)[10-12]. PD affects 0.3% of the worldwide population, with prevalence increasing by age; in fact, it is estimated to be 1% in people over 60 years of age and 3%-5% in individuals over 85[13,14]. The clinical features of PD include typical motor symptomatology (bradykinesia, resting tremor, postural instability, and gait difficulties) and non-motor symptoms (dysautonomia, sleep disturbances, mood, and cognitive disorders)[15,16].
T2D, AD and PD are all characterized by a multifactorial etiology, involving the interplay among genetic, epigenetic and environmental factors[17,18]. Interestingly,there are lines of evidence at the epidemiological, cognitive and neuropathological levels that seem to link T2D to AD and PD[19]. In particular, brain insulin resistance could represent the bridge linking metabolic disorders to neurodegenerative/movement pathological conditions[20]. Insulin is transported via the blood brain barrier to the central nervous system, where it regulates local blood and cerebrospinal fluid glucose levels. Nevertheless, it is thought that the principal activity in the brain may be related to the regulation of synaptic plasticity and cognitive functions[7,21].Moreover, a little proportion of insulin may be produced in the brain, as well. Indeed,insulin levels detected in humans and rodents have been found to be lower than those in the systemic circulation. However, differences in the levels of insulin among AD brains and age-matched controls have not been established[7]. Insulin Receptors (IRs)are well distributed in the brain, especially in the cortex, hippocampus and hypothalamus, corroborating the importance of brain insulin signaling[7,21]. The“diabetic brain” may suffer of the hyperglycemia and insulin resistance arising from the decrease in insulin receptor expression or activity[7,21]. This alteration may lead to the activation of pathogenic processes, namely enhanced production of reactive oxygen species (ROS) and pro-inflammatory cytokines, that trigger inflammatory responses also in the brain, advanced glycation products and dysfunctions of autophagic functions. Moreover, insulin resistance is able to increase the production and secretion of beta amyloid (Aβ) and alter the molecular pathways involved in the phosphorylation of Tau protein: both Aβ and hyperphosphorilated-Tau are known to misfold, aggregate and accumulate leading to the loss of synapses and death of neurons, which are typical of neurodegeneration processes[7,22-28]. Indeed, the increased neuroinflammation represents a pathological feature shared by all of the three agerelated pathologies[29]. Thus, the existence of common triggering factors reflects the contribution of mutual genetic and epigenetic features in the etiopathogenetic mechanisms underlying AD, PD and T2D. On this subject, this review will summarize the shared (epi)genomic features that characterize these complex pathologies.
SHARED GENETIC MAKE-UP AND FUNCTIONAL PATHWAYS AMONG T2D, AD AND PD
Several studies have attempted to dissect the contributing genetic background(s) to determine the susceptibility to T2D, AD and PD. Concerning T2D, most of the identified genetic risk factors are mainly involved in the maintenance of β-cell homeostasis and in the modulation of insulin metabolism[2,30,31]. As previously mentioned, insulin resistance has been reported to likely influence brain functions and neuronal activity. Concerning the genetic susceptibility factors of AD and PD,several genome-wide association studies (commonly referred to as GWAS) have identified many genetic polymorphisms associated with the onset and progression of sporadic forms of AD and PD. Most of them have been found to be located within genes involved in dopamine metabolic process, apoptosis, autophagy-related pathways, Aβ cascade, Tau pathology, neuroinflammation, regulation of neuronal transmission, and survival[17,32-35]. The availability of GWAS and bioinformatic approaches has allowed for the identification of 927 single nucleotide polymorphisms(SNPs) associated with both T2D and AD in populations of European ancestry.Intriguingly, 395 of these SNPs have been reported to share the same risk allele between T2D and AD[36]. These SNPs are involved in immunity/inflammation-related pathways, cell-cell communication and neuronal plasticity, whose dysregulation may lead to increase in the neuroinflammation typically occurring in T2D and AD[7,37,38].Polymorphisms within theIDE/HHEXregion have also been investigated as combined susceptibility factors for T2D and AD (Table 1)[39-41]. Notably,IDEcodes for the enzyme responsible for insulin clearance, although it is also able to degrade Aβ peptide in neurons and glia cells[7,39].
A recent study performed on populations of European ancestry has described the association of 14 common SNPs with both T2D and AD; these are located inTP53INP1,NDUFAF6,TOMM40,BTBD16,PLEKHA1,PVRL2andAPOC1genes[42,43](Table 1). Interestingly, these genes encode proteins involved in the regulation of autophagy, apoptosis, response to oxidative stress, mitochondrial function and lipid metabolism, and their overall dysregulation can contribute to the etiopathogenetic pathways underlying T2D and AD[7,22]. Of note, Haoet al[34], 2015 and Wanget al[39],2017 found that both disorders shared the same risk variant in SNPs (rs10510109 and rs2421016) located inBTBD16andPLEKHA1genes (Table 1). This is of particular interest, as different SNPs withinPLEKHA1have been associated with age-related macular degeneration (an ocular neurodegenerative complex disease)[44-46]and they map on the 10q26.13 locus, which also contains another age-related macular degeneration-associated gene (ARMS2/HTRA1)[47,48]. Given these data, the genetic architecture of the 10q26.13 region may be investigated for its potential contribution to neurodegeneration and could be addressed as a shared susceptibility locus for T2D and AD. Moreover, the presence of shared genetic polymorphisms associated with both diseases may also be exploited to predict the risk of developing AD in individuals already suffering from T2D.
Less information is available concerning the genetic overlap between T2D and PD.The possible link between T2D-associated genetic loci and AD/PD has beeninvestigated in a study involving 500 PD and 400 AD patients of Asian ancestry. The authors reported four SNPs located inCDC123andCDKN2Bgenes mutually associated with T2D and PD. However, this association was not confirmed after correction for multiple testing[49](Table 1).CDC123andCDKN2Bexert a role in cell
cycle regulation, and their dysfunction leads to alterations in cell homeostasis,suggesting that the genetic association with T2D and PD should be further investigated in larger cohorts and different populations. Furthermore, four different genes, namelyKANSL1,CXCR4,MAP3K14andCRHR1, were found to be shared between PD and type 1 diabetes in a study aiming to evaluate the common risk factors between PD and autoimmune disorders[50](Table 1). Intriguingly,CXCR4andMAP3K14are involved in the regulation of neuronal inflammatory responses. In particular,CXCR4is involved in microglia recruitment, neuronal guidance and neurodevelopmental processes[51], whereasMAP3K14mediates NFκB signaling(involved in immunological cytotoxicity) in brain neurons[52]. Moreover, the CXCR4 protein has been found to be overexpressed in a rodent model of diabetic neuropathic pain[53].KANSL1has been found to be associated with AD, thus suggesting that the encoded protein may take part in neuronal development. Indeed, KANSL1, as part of the NLS1 complex which regulates histone acetylation, is mainly involved in the epigenetic regulation of chromatin[54]. Interestingly, mutations withinKANSL1are able to cause intellectual disability and developmental delay[54,55].CRHR1is known to be involved in the activation of hypothalamic-pituitary-adrenal axis, leading to secretion of cortisol that, in turn, causes insulin resistance. Notably, the chronic stress activated by the adrenal secretion of cortisol represents a risk factor for AD onset and progression[56]. Given these lines of evidence, the association between this set of genes and their potential involvement in etiopathogenetic pathways leading to T2D, AD and PD should be further elucidated.
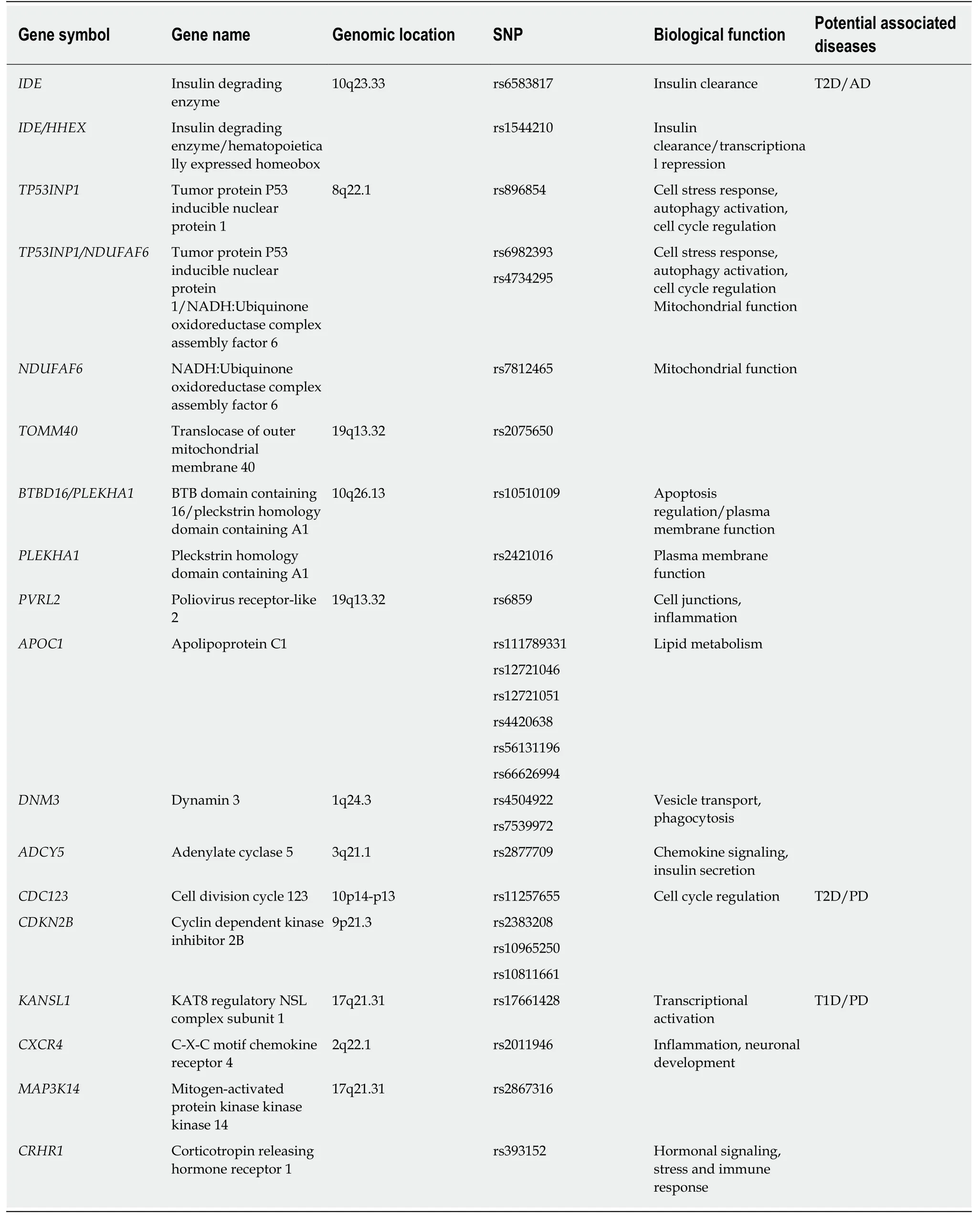
Table 1 Subset of genetic variants and genes found to be associated with type 2 diabetes, Alzheimer's disease and Parkinson's disease,as well as those associated with type 1 diabetes and Parkinson's disease[36,39,41-43,49,50]; Biological functions have been obtained from literature data[7,22,39,49,51-56] and GeneCards (https://www.genecards.org)
In addition to the identification of shared genetic variants, the investigation of common gene expression profiles may facilitate the discovery and exploration of molecular pathways that are deregulated in T2D, AD and PD. On this subject,Rahmanet al[57], 2018 reported intriguing insights, exploiting human gene expression datasets. Among the significant Gene Ontologies (known as GOs) and Kyoto Encyclopedia Genes and Genomes (known as KEGG) pathways shared by T2D and AD, pathways involved in glycosphingolipid biosynthesis, immune/inflammatory response, regulation of neurotransmitter transports, synaptic vesicle formation, lipid metabolism and apoptosis have been identified. T2D and PD share genes involved in immune-related networks, cell adhesion, mitochondrial activity, connective tissue/extracellular matrix organization, and synaptic maturation. Indeed,neuroinflammation may represent a common hallmark among T2D, AD and PD,given that most of the shared genes are implicated in the regulation of inflammatory networks. Interestingly, Santiagoet al[58], 2013 found thatAPPmRNA was overexpressed in the whole blood of both T2D and PD patients. Therefore, the knowledge of shared genetic factors and gene expression profiles may help to further dissect the molecular network characterizing and linking T2D, AD and PD (Figure 1).
INSIGHTS INTO COMMON EPIGENETIC BACKGROUND(S)OF T2D, AD AND PD
The human genome is able to dynamically interact with the environment through epigenetic modifications, which altogether create the complex machinery designated to regulate lifetime and aging processes. In fact, epigenetics modulate gene expression without altering the DNA sequence. This is possible by means of different kinds of epigenetic modifications, including DNA methylation and histone modifications(which might affect gene transcription), and noncoding (nc)RNAs (which might change gene expression at the post-transcriptional level)[59]. Given the crucial role of epigenetics in the modulation of gene expression, its alteration can contribute to pathogenesis and progression of several age-related diseases, including metabolic,neurodegenerative and movement disorders[17,60]. The existence of a shared epigenetic background among T2D and neurodegenerative diseases deserves to be investigated.As a matter of fact, the gene expression signatures shared among T2D and AD/PD[57,58]may also be related to the presence of common epigenetic alterations[61]. On this subject, there are intriguing hypotheses that could be evaluated. For instance, the analysis of long-range chromatin contacts among regulatory regions and their target genes will provide insights into how epigenetic background(s) may modify chromatin conformation[62]and thus gene expression profiles in the context of T2D, AD and PD.Moreover, an interaction between micro (mi)RNA-661 andBACE1mRNA was found to cause a reduced expression of the resultant protein in pancreatic islets and contribute, thereby, to the development of T2D[63]. Of note,BACE1is involved not only in the regulation of insulin biogenesis but also in the formation of Aβ so that it could be also investigated in the etiopathogenesis of AD[64].
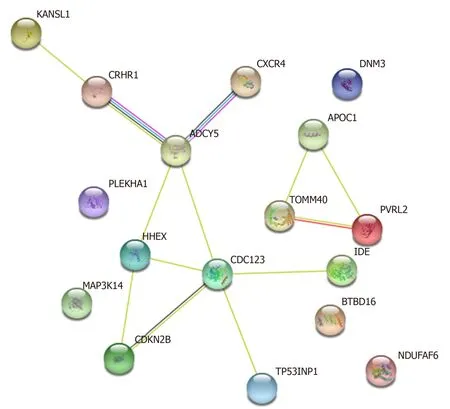
Figure 1 Known interaction networks among the potentially shared genes.
Furthermore, sirtuins are a family of histone deacetylases, playing critical roles in the physiology of metabolism, central nervous system, and immune system. In fact,these epigenetic modifiers are involved in a variety of molecular pathways underlying different complex diseases (cancer, diabetes, and neurodegenerative disorders)[65]. Given their role, sirtuins may be addressed as potential therapeutic targets able to counteract the progression of T2D, AD and PD through their epigenetic activity[66].
The study of DNA methylation affecting mitochondrial genes could unveil interesting insights into the pathogenesis of T2D, AD and PD. In fact, alteration of DNA methylation status has been supposed to be responsible for the reduction of complex I and IV subunits in AD and PD human brain samples[67]. Moreover, it has been demonstrated that the alteration of miR-181a/b levels impacts mitochondrial biogenesis and turnover in the brain, through the modulation of autophagy and mitophagy-related pathways[68]. These miRNAs could be, therefore, investigated for their potential role in the common pathogenetic processes leading to T2D and AD/PD. Furthermore, the study of other miRNAs and ncRNAs related to these disorders could be helpful for designing innovative class of drugs (epidrugs).
CONCLUSION
A growing body of evidence suggests the existence of multilevel networks of pathogenetic pathways which mutually contribute to the onset and progression of metabolic, neurodegenerative and movement disorders. However, few shared genetic contributors have been well characterized and a common epigenetic landscape needs to be explored. Of note, in T2D, an impairment of glucose metabolism in brain generates oxidative stress, leading to the alteration of autophagy-related pathways,mitochondrial dysfunction, increased neuronal apoptosis and, eventually, depletion of synapsis[7]. Overall, these alterations contribute to the formation of amyloid plaques and neurofibrillary tangles in AD and to the deterioration of dopaminergic neurons in different brain regions in PD[1]. As mentioned, despite a plethora of data highlighting a possible overlapping of disease mechanisms involved in T2D, AD and PD, the critical molecular and genetic features remain to be clarified. The genetic polymorphisms (Table 1) shared with T2D, AD and PD are located within genes involved not only in brain insulin signaling but also in neuroinflammation-related pathways[34-52]. This evidence is also corroborated by the expression data obtained by the investigation of human patients and animal models presenting these pathologies[57].
Understanding the contribution of genetics, epigenetics and environment in determining the susceptibility, onset and progression of T2D, AD and PD will be crucial to achieve a deeper knowledge of metabolic, neurodegenerative and movement disorders. On this subject, the enhancement of social and cognitive activities in the high-income countries seems to strengthen the resilience against neurodegeneration, leading to a stable or reduced incidence of dementia in these regions[69,70]. On the other hand, T2D prevalence is also rising in the more developed areas[71]. Given this data and considering that T2D is regarded overall as a risk factor for dementia[72], the contribution of T2D to the development of neurodegeneration needs to be monitored. Moreover, these lines of evidence encourage the further exploration of gene-environment interactions in order to understand the similarities and the differences in the etiopathogenesis underlying AD, PD and T2D. Indeed,more comprehensive and higher resolution (epi)genomic studies should be implemented in order to collect information on genome architecture, DNA methylation, histone modifications, ncRNAs and three-dimensional genome organization. These large-scale data should be exploited to integrate genomic,epigenomic, transcriptomic, metabolomic and proteomic information with the clinical phenotype and draw the network of interactions which build up a “diseaseinteractome”.
Indeed, the fine knowledge of the disease interactome could highlight the molecular relationships existing among T2D, AD, PD which, thereby, could be exploited to treat these conditions through a network medicine approach, able to integrate all these interactions to understand the molecular and cellular perturbations underlying diseases, providing insights and targets for the accurate diagnosis and treatment[73]. By this way, the patient could benefit from a healthcare approach based on a multilevel characterization of his condition, derived not only by clinical and molecular testing but also by his environmental and social backgrounds.
杂志排行

World Journal of Diabetes的其它文章
- Role of sodium-glucose co-transporter-2 inhibitors in the management of heart failure in patients with diabetes mellitus
- Severity of the metabolic syndrome as a predictor of prediabetes and type 2 diabetes in first degree relatives of type 2 diabetic patients: A 15-year prospective cohort study
- Evaluation of oxidative stress levels in obesity and diabetes by the free oxygen radical test and free oxygen radical defence assays and correlations with anthropometric and laboratory parameters
- Maternal low protein diet induces persistent expression changes in metabolic genes in male rats
- Fundamentals about onset and progressive disease character of type 2 diabetes mellitus