Ce + XF3(X = N,P和As)反应产物的量子化学研究
2020-06-30李宇情郭雅洁黄正国
李宇情,郭雅洁,黄正国
Ce + XF3(X = N,P和As)反应产物的量子化学研究
李宇情,郭雅洁,黄正国
(天津师范大学 化学学院,天津 300387)
采用ab-initio方法研究了Ce+XF3(X=N,P和As)反应的可能产物(CeXF3、XCeF3、FXCeF2和F2XCeF)的平衡几何结构和能量,并使用AIM、自然布居电荷(NPA)、Pipek-Mezey定域化分子轨道和FBO分析研究了这些产物分子中的成键性质。结果表明,所有分子的基态自旋多重度均为三重态。对于XCeF3,FXCeF2和F2XCeF,氮化物的热力学稳定性最高,其次是砷化物,磷化物的热力学稳定性最低;CeXF3的情况则正好相反。AIM分析表明,大多数Ce-X、X-F和Ce-F键主要呈现闭壳层相互作用特征,且具有部分共价作用特征。由于F2XCeF分子中存在agostic作用,Ce-F1(或F2)键表现出闭壳层相互作用的特征;且RCP与BCP相近,表明X-Ce-F1(或F2)三元环的稳定性较弱。
电子结构;从头算;分子中的原子理论(AIM);agostic作用
常见的第一完整行中元素C,N和O可以用2p价轨道形成多重键,这些键决定了许多简单化合物(如HC≡CH,N≡N和O=O)的化学性质。然而元素周期表中第二、第三和第四个完整行中的主族元素不倾向于形成多重键。尽管p-区元素可以形成双键,但很少发现三重键的形成。具有多重键的f区元素(镧系元素和锕系元素)并不常见,并且人们在理论计算[1-6]和实验合成[1-5]上为寻找具有两个锕系金属之间以及锕系元素和主族元素之间的多重键化合物做出了很大的努力。Andrews和王雪峰使用激光烧蚀U原子首次制备了亚甲基铀和次甲基铀化合物,包括CH2=UH2,CH2=UHF,CH2=UF2,HC≡UF3和FC≡UF3[7-9]。人们还通过激光烧蚀U原子与CHF3,NF3和PF3反应制备了HC≡UF3,N≡UF3和PtUF3分子,并分析了它们的化学键性质[10-11]。在激光烧蚀或紫外光激发下Mo和W原子与AsF3发生反应,可形成稳定的三角型砷化物As≡MF3[12]。人们利用基质隔离红外光谱技术对含有三重键的铀化合物进行了研究,制备了NUN和CUO分子,并用质谱法检测到的[NUO]+阳离子[13-16]。Andrews和Cho使用激光烧蚀的Th和U与CH4及NH3反应制备最简单的锕系亚甲基和亚胺分子CH2=AnCH2和HN=AnH2,并用基质红外光谱和密度泛函理论(DFT)计算进行了表征[7,17-20]。使用U与氟类似物NF3,PF3和AsF3反应产生了稳定的U(VI)产物N≡UF3、P≡UF3和As≡UF3,并且使用DFT来进一步表征这些简单的末端氮化物,磷化物和砷化物[11,21]。由此可见,f区元素通过多重键形成的化合物具有巨大的研究价值,本文拟对Ce + XF3(X = N,P和As)反应产物进行量子化学理论研究,希望能够获得对锕系元素化合物多重键性质的新认识。
1 计算方法
采用从头算方法计算XCeF3、FXCeF2、F2XCeF和CeXF3(X = N,P和As)分子的结构、能量和光谱性质,所有计算均使用Gaussian09程序完成[22]。考虑到重元素的相对论效应,对于Ce使用Stuttgart-Dresden相对论有效核势(RECP)及其相应的基组,且保留30个电子[23],其它元素均使用Def2TZVPP基组[24-25]。首先,在没有任何对称限制下用MP2(full)方法对XCeF3、FXCeF2、F2XCeF和CeXF3(X = N,P和As)分子进行优化,然后在同一计算水平上采用解析二阶导数方法计算这些分子的谐振频率,以确认优化得到的分子构型是能量极小值并计算零点振动能(ZPVE)。最后,使用相同的RECP和基组对已优化好的分子结构计算CCSD(T)单点能,以获得更精确可靠的能量。
采用Bader的分子中原子(AIM)方法[26-27]和模糊键级(FBO)方法[28-29]对XCeF3、FXCeF2、F2XCeF和CeXF3(X = N,P和As)的化学键性质进行研究。在进行AIM分析和ELF分析时,对于Ce原子采用WTBS全电子基组来生成波函数[30-31]。FBO和AIM计算使用Multiwfn完成[32]。
2 结果和讨论
2.1 结构
XCeF3、FXCeF2、F2XCeF和CeXF3(X = N,P和As)分子可能的电子自旋多重态的结果表明,所有分子的基态自旋多重度均为三重态,图1示出了在MP2(full)计算水平上获得的所有基态分子的结构参数。
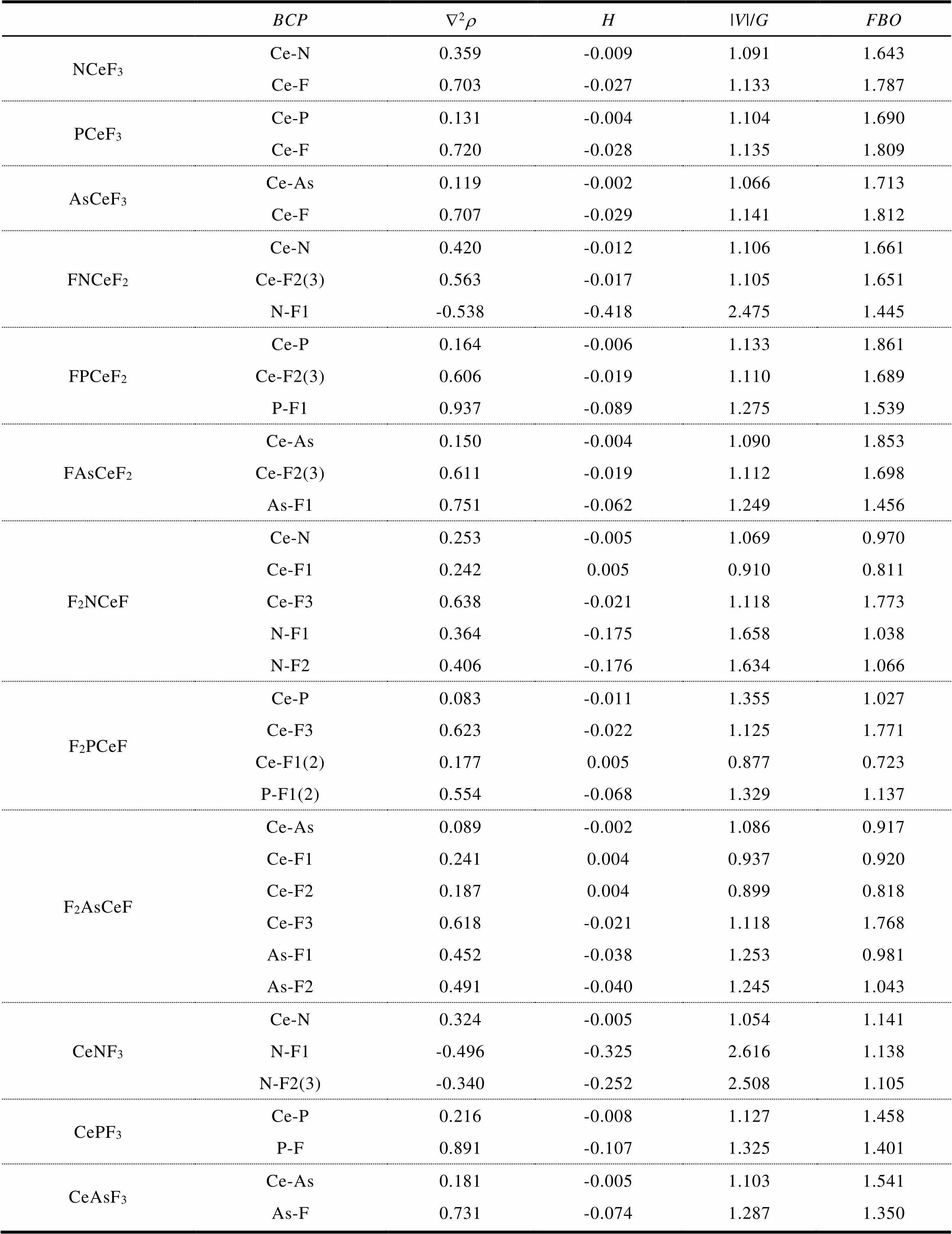
如图1所示,所有XCeF3分子属于C3v点群,对称性最高。CePF3和CeAsF3分子也具有C3v对称性,而CeNF3属于Cs点群。FXCeF2均为具有C2v对称性的平面分子,其中F1、X和Ce共线。F2XCeF(X = N和As)属于对称性最低的C1点群,其中F3- Ce-X-F2二面角接近180°,而F2PCeF分子的对称性不同于F2XCeF(X = N和As),属于Cs点群。F2XCeF(X = N,P和As)中ÐCe-X-F1(F2)均为锐角,且该角度随着X由N到As而逐渐减小,这主要是因为F2XCeF分子中形成了agostic作用的缘故。随着XCeF3分子中的第一个F原子由Ce迁移到X原子上,Ce-X键变短,可见Ce-X键有变强的趋势;而当第一个F原子由Ce迁移到X原子上后,由于F2XCeF分子中存在agostic作用,所以Ce-X键反而变长。同样,受agostic作用的影响,F2XCeF分子中的X-F键比FXCeF2的X-F键要长。对于含不同X原子的XCeF3分子,随着X由N到As,X-Ce键长逐渐变长;在FXCeF2、F2XCeF和CeXF3分子中也有类似的规律,即RN-Ce< RP-Ce dA-B=A-B-A-B(1) 其中A-B是A-B键的键长,A和B分别是A和B原子的共价单键半径[33]。dA-B越小,A-B化学键越强,反之亦然。计算出的所有分子中的dX-Ce、dX-F同样示于图1。图1中,FXCeF2中所有X-Ce键的dRX-Ce均为负值,说明这些键应该强于相应的典型X-Ce单键;与之相反,F2XCeF中X-Ce键的dRX-Ce均为正值意味着这些键应该弱于相应的X-Ce单键。在XCeF3和FXCeF2中,P-Ce键最长而N-Ce键最短,说明P-Ce键最弱而N-Ce键最强;FXCeF2中X从N变到As,X-Ce键长逐渐减小,而F2XCeF中随着X从N变到As,X-Ce键的dRX-Ce逐渐增大,说明F2XCeF中X-Ce键逐渐减弱,这种情况可部分归因于agostic作用。比较FXCeF2和F2XCeF中的dX-F可知,由FXCeF2到F2XCeF,dX-F变大,所以X-F键变弱,这同样可归因于agostic作用。在CeXF3分子中,随着X从N变到As,X-Ce键的dRX-Ce逐渐减小,说明X-Ce键是逐渐增强的。应该说明的是,文献[33]中的共价半径数据并不是专门为本研究体系所准备的,并不一定能够精确反映X-Ce和X-F键的强弱,所以通过dX-Ce(或dX-F)仅仅可以粗略地估计X-Ce(或X-F)键的强度,后面将会对这些化学键的性质做进一步的分析。 图1 在MP2(full)/Def2TZVPP水平上计算的XCeF3,FXCeF2,F2XCeF和XCeF3(X = N,P和As)的结构参数(键长单位为Å,键角和二面角单位为度)。括号内数值为根据公式(1)计算的dRX-Ce(或dRX-F) 在优化好的结构基础上,使用CCSD(T)方法计算CeXF3、XCeF3、FXCeF2和F2XCeF分子相比于反应物Ce + XF3(X = N,P和As)的相对能量,由于CCSD(T)非常耗时,所以CCSD(T)计算没有考虑零点能校正(ZPVE),计算结果列于表1。由表1可知,在这四类分子中,CeXF3的D最大,说明Ce-X键能较小,且CeXF3相比于Ce + XF3的热力学稳定性最低,特别是CeNF3的D为正值,表现出热力学上的不稳定性;热力学稳定性次低者是F2XCeF分子。在所有氮化物中,NCeF3的热力学稳定性最高;而对于磷化物和砷化物,热力学稳定性最高的则是FXCeF2。同时,CeXF3中的F原子在热力学上有由X原子迁移到Ce原子上的趋势。 表1 在CCSD(T)/Def2TZVPP水平上计算的XCeF3,FXCeF2,F2XCeF和XCeF3(X = N,P和As)相比于Ce + XF3的相对能量DE(单位:kcal·mol-1)a 注:aCCSD(T)能量没有进行ZPVE校正。 由表1可知,对于XCeF3分子,NCeF3的DE最小,其次是AsCeF3,而PCeF3的DE最大,说明XCeF3分子的热力学稳定性次序为 NCeF3> AsCeF3> PCeF3。 对于FXCeF2和F2XCeF分子,也有类似的规律;CeXF3的情况则正好相反,CeNF3的热力学稳定性最低,CeAsF3次之,而CePF3的热力学稳定性最高。综上分析,CeNF3中的F原子发生由N原子向Ce原子上迁移的热力学趋势最强,其次是CeAsF3,而CePF3发生F原子迁移的热力学趋势最弱。 CeXF3、XCeF3、FXCeF2和F2XCeF分子(X = N,P和As)的AIM分析结果列于表2,这些分子中电子密度的拉普拉斯值(Ñ2r)等值线图见图2和图3。 表2 使用MP2(full)方法及全电子相对论基组计算的XCeF3,FXCeF2,F2XCeF和XCeF3(X = N,P和As)的AIM参数和模糊键级(FBO) 如图2所示,在XCeF3分子中,Ce-X的键临界点(BCP)处于电荷耗散区域(实等值线),表明Ce-X键呈现出闭壳层相互作用特征;且XCeF3(X = N和P)分子中X原子周围有一个电荷聚集区域(虚等值线),所以Ce-N(或Ce-P)键具有部分共价作用特征;AsCeF3分子中As原子只有在朝向Ce原子的方向存在一个电荷聚集区域,所以Ce-As键也具有部分共价作用特征,但相对较弱,可见由Ce®As的电荷转移程度弱于Ce®N(或P)的电荷转移程度。在XCeF3分子的Ce-F键中也有类似的现象,即Ce-F键表现出闭壳层相互作用的特征,并且电荷从Ce原子转移到了X原子和F原子上。 同样,通过分析FXCeF2分子的Ñ2r等值线图可知,在FXCeF2分子的Ce-X和Ce-F键均呈现出闭壳层相互作用的特征;且FXCeF2(X= N和P)分子中X原子周围在朝向Ce原子的方向有一个电荷聚集区域,所以Ce-N(或Ce-P)键具有部分共价作用特征;而FAsCeF2分子中在Ce-As键上没有发现电荷聚集区域,所以As原子由Ce原子上获得电子的能力弱于N(或P)原子。在FNCeF2分子中N-F键BCP处于电荷聚集区域,所以表现为共价作用特征;在FXCeF2(X= P和As)分子中X-F键BCP处于电荷耗散区域,表现为闭壳层作用特征。 由CeXF3分子的Ñ2r等值线图可知,Ce-X键的BCP处于电荷耗散区域,呈现出闭壳层相互作用的特征。同时X原子周围在朝向Ce原子的方向有一个电荷聚集区域,所以Ce-X键也具有部分共价作用特征。同FXCeF2分子类似,在CeNF3分子中N-F键BCP处于电荷聚集区域,表现为共价作用特征,FXCeF2(X= P和As)分子中X-F键BCP处于电荷耗散区域,表现为闭壳层作用特征。如前所述,F2XCeF分子的对称性较低,且存在agostic作用,所以本文选择了F3-Ce-X(plane 1)、Ce-X-F1(plane 2)和F1-X-F2(plane 3)这三个平面做F2XCeF分子的Ñ2r等值线图,示于图3。 图2 XCeF3,FXCeF2和XCeF3(X = N,P和As)的Ñ2r(r)等值线图。其中,虚线表示电荷聚集区(Ñ2r(r)< 0),实线表示电荷耗散区(Ñ2r(r)> 0);连接原子核的棕色粗实线表示键径,将不同原子核割开的蓝色粗实线表示分子面内的零流面,键径与零流面的交点为键临界点(BCP),图3同。 图3 F2XCeF(X = N,P和As)的Ñ2r(r)等值线图 如图3的plane 1所示,与FXCeF2分子的Ce-X键类似,F2XCeF分子的Ce-X键均呈现闭壳层相互作用特征,且F2XCeF(X = N和P)分子中X原子周围沿Ce-X键径方向有电荷聚集区域,所以Ce-N(或Ce-P)键具有部分共价作用特征,而FAsCeF2分子中沿Ce-As键径方向没有发现电荷聚集区域,这是因为As原子由Ce原子上获得电子的能力弱于N(或P)原子。由于F2XCeF分子中存在agostic作用,所以F1、F2原子在与X原子成键的同时,还与Ce原子成键。由图3的plane 2可知,Ce-F1(或F2)键表现出闭壳层相互作用的特征,且环临界点(RCP)与BCP相近,表明X-Ce-F1(或F2)三元环的稳定性较弱。由图3的plane 3可知,X原子与F1、F2原子成键,主要呈现出闭壳层相互作用的特征,同时兼具部分共价作用特征,且X原子周围的电荷聚集区域按照N®P®As趋势减少。 如表2所示,所有分子中X-Ce键的BCP的Ñ2r均为正值,总能量密度(H)均为负值,因此X-Ce键表现出部分共价键特征,同时此处的|V|G/值均在1.0~1.4范围内,进一步证明X-Ce表现出部分共价作用[34-37]。由于FNCeF2和CeNF3中N-F键BCP的Ñ2r和H均为负值,所以这些N-F键呈现为共价作用,同时它们的|V|G/值均大于2.4,进一步确认了它们的共价作用特征,这与前面关于Ñ2r等值线图的分析一致。除此之外的X-F(X= N,P和As)键均主要表现为闭壳层相互作用特征,兼具部分共价作用特征,且与相应的Ce-X键相比,它们的H值更负,而|V|G/值则更大,所以这些X-F键比相应的Ce-X键具有更多的共价作用特征[34]。 类似地,由表2可知,在XCeF3和FNCeF2(X= N,P和As)中,Ce-F键均主要表现为闭壳层相互作用特征,兼具部分共价作用特征。F2NCeF分子中的Ce-F3与此类似,而由于F2NCeF中的F1、F2参与形成了agostic作用,所以Ce-F1(或Ce-F2)键表现出了与Ce-F3键不同的特点。Ce-F1(或Ce-F2)键BCP的Ñ2r和H均为正值,表现为闭壳层相互作用特征,同时它们的|V|G/值均小于1.0,进一步确认了它们的闭壳层相互作用特征。 使用MP2(full)方法计算XCeF3、FXCeF2和F2XCeF(X= N,P和As)分子FBO的结果也列于表2。如表2所示,XCeF3中X-Ce键的FBO由X从N变到As逐渐增大,似乎As-Ce键强一些而N-Ce键稍微弱,而FXCeF2和F2XCeF的FBO顺序为P-Ce>As-Ce>N-Ce,P-Ce键似乎比其他二者稳定,而N-Ce键最弱,这与前面的结构、能量分析的结果有些出入,一个可能的解释是用FBO键级来描述X-Ce键并不非常完美。 表3为XCeF3、FXCeF2、F2XCeF和CeXF3(X = N,P和As)中各原子的自然电荷。 如表3所示,所有分子中Ce原子均带正电,说明发生了由Ce原子向其它原子或基团的电子转移。在CeXF3分子中,尽管X原子的电荷数均为正值,XF3基团整体仍然带负电荷,所以在CeXF3分子中存在Ce®XF3的电荷转移,且电荷转移程度最高的是CeNF3,其次是CeAsF3,CePF3中的Ce®XF3电荷转移程度最低。 在F2XCeF分子中,F3原子由CeXF3中的X原子迁移到Ce原子上形成Ce-F键,由于X原子少了一个F原子成键,所以尽管F2XCeF中X原子电荷仍然呈正值,但比CeXF3中X的电荷数明显减小;与此相对应的是,F2XCeF中的XF2基团比CeXF3中XF3基团带了更多的负电荷,且F2XCeF中发生Ce®XF2电荷转移程度最高的是F2NCeF,其次是F2AsCeF,F2PCeF中的Ce®XF2电荷转移程度最低,这种次序与上述CeXF3中的Ce®XF3电荷转移次序类似,同时这种次序与Ce原子上电荷数值大小次序也是一致的。 在FXCeF2分子中,由于X原子只与F1形成X-F1键,而F2和F3原子由CeXF3中的X原子迁移到Ce原子上形成两个Ce-F键,所以导致X原子上的电荷数进一步减小,其中P和As原子的电荷数为正值,只有电负性与F原子较为接近的N原子的电荷数为负值。此外,FXCeF2分子中发生Ce®XF电荷转移的次序为FNCeF2> FAsCeF2> FPCeF2,这种次序与上述Ce®XF3(CeXF3)以及Ce®XF2(F2XCeF)电荷转移次序是一致的,同时也与Ce原子上电荷数值大小次序也是一致的。 表3 在MP2(full)/Def2TZVPP水平上计算的XCeF3,FXCeF2,F2XceF和XCeF3(X = N,P和As)的自然电荷 在XCeF3中,由于X原子不再与电负性更强的F原子成键,所以X原子的电荷均呈负值,且电荷数绝对值从N到As逐渐减小,同时伴随着Ce原子上电荷数的递减,这与X原子的电负性次序一致,同时说明XCeF3中X原子从Ce原子上获得电子的能力随着X原子电负性的减小而逐渐减弱。此外,XCeF3中Ce原子的电荷数大于FXCeF2、F2XCeF和CeXF3分子中Ce原子的电荷数,说明XCeF3中电荷转移程度更高,且多数电荷转移给了电负性最高的F原子。 图4和图5分别为XCeF3和FXCeF2(X = N,P和As)分子的部分分子轨道图。 从图中可以看出,X的孤对电子以及Ce附近定域程度最高,XCeF3中X-Ce键周围有一个σ键和两个单占据的π键,σ键呈环形环绕在X-Ce键周围,而两个π键以互相垂直的方向分布在X- Ce键两侧,其中π键只有α轨道上有电子,而β轨道上无电子,是典型的单占据π键,因此XCeF3中X-Ce键可认为是特殊的三键。FXCeF2中X-Ce周围有一个两电子π键和一个单电子π键,这样看起来似乎FXCeF2中X-Ce比XCeF3中X-Ce键更强,但是在FXCeF2中,X-Ce键中的σ轨道更像X上的孤电子对轨道,正因为这个原因,才使得FXCeF2中X-Ce键较XCeF3中X-Ce键弱。 图4 在MP2/Def2TZVPP水平上计算的XCeF3(X = N,P和As)的Pipek-Mezey定域化轨道 图5 在MP2/Def2TZVPP水平上计算的FXCeF2(X = N,P和As)的Pipek-Mezey定域化轨道 本文采用MP2(full)和CCSD(T)方法研究了Ce+XF3(X=N,P和As)反应的可能产物(CeXF3、XCeF3、FXCeF2和F2XCeF),主要结论如下: 所有分子的基态自旋多重度均为三重态。所有XCeF3分子属于C3v点群,CePF3和CeAsF3分子也具有C3v对称性,而CeNF3属于Cs点群。FXCeF2均具有C2v对称性,F2XCeF(X = N和As)属于对称性最低的C1点群,而F2PCeF分子的属于Cs点群。在F2XCeF分子中存在agostic作用。 CeXF3中的F原子在热力学上有由X原子迁移到Ce原子上的趋势,所以CeXF3的热力学稳定性最低,其次是F2XCeF分子。在所有氮化物中,NCeF3的热力学稳定性最高,对于磷化物和砷化物,热力学稳定性最高的则是FXCeF2。XCeF3分子的热力学稳定性次序为NCeF3> AsCeF3>PCeF3。对于FXCeF2和F2XCeF分子,也有类似的规律,而CeXF3的情况与上述分子正好相反。所以,CeNF3中的F原子发生由N原子向Ce原子上迁移的热力学趋势最强,其次是CeAsF3,而CePF3发生F原子迁移的热力学趋势最弱。 在所有分子中,大多数Ce-X、X-F和Ce-F键主要呈现出闭壳层相互作用特征,且具有部分共价作用特征。由于F2XCeF分子中存在agostic作用,Ce-F1(或F2)键表现出闭壳层相互作用的特征,且RCP与BCP相近,表明X-Ce-F1(或F2)三元环的稳定性较弱。 在所有分子中均发生了由Ce原子向其它原子或基团的电子转移。在CeXF3分子中的Ce®XF3电荷转移程度由高到低的次序是CeNF3> CeAsF3>CePF3。与此类似的是F2XCeF中发生Ce®XF2电荷转移的次序(F2NCeF> F2AsCeF> F2PCeF)以及FXCeF2分子中发生Ce®XF电荷转移的次序(FNCeF2>FAsCeF2>FPCeF2)。XCeF3中电荷转移程度更高,且多数电荷转移给了电负性最高的F原子。 致谢:本课题得到天津市功能分子结构与性能重点实验室资金资助,特此感谢! [1] Straka M, Pyykko P. Linear HThThH: A candidate for a Th-Th triple bond[J]. J Am Chem Soc, 2005, 127(38): 13090-13091. [2] Gagliardi L, Roos BO. Quantum chemical calculations show that the uranium molecule U-2 has a quintuple bond[J]. Nature, 2005, 433(7028): 848-851. [3] Roos BO, Gagliardi L. Quantum chemistry predicts multiply bonded diuranium compounds to be stable[J]. Inorg Chem, 2006, 45(2): 803-807. [4] Cavigliasso G, Kaltsoyannis N. On the paucity of molecular actinide complexes with unsupported metal- metal bonds: A comparative investigation of the elec- tronic structure and metal-metal bonding in U(2)X(6) (X = Cl, F, OH, NH(2), CH(3)) complexes and d-block analogues[J]. Inorg Chem, 2006, 45(17): 6828-6839. [5] Frenking G, Tonner R. Theoretical chemistry - The six- bond bound[J]. Nature, 2007, 446(7133): 276-277. [6] Ephritikhine M. The vitality of uranium molecular chemistry at the dawn of the XXIst century[J]. Dalton Trans, 2006, (21): 2501-2516. [7] Lyon JT, Andrews L, Malmqvist P-A, et al. Infrared spectrum and bonding in uranium methylidene di- hydride, CH2=UH2[J]. Inorg Chem, 2007, 46(12): 4917- 4925. [8] Lyon JT, Andrews L. Formation and characterization of the uranium methylidene complexes CH2=UHX (X = F, C, and Br)[J]. Inorg. Chem, 2006, 45(4): 1847-1852. [9] Lyon JT, Andrews L, Hu H-S, Li J. Infrared spectra and electronic structures of agostic uranium methylidene molecules[J]. Inorg Chem, 2008, 47(5): 1435-1442. [10] Lyon JT, Hu H-S, Andrews L, Li J. Formation of unprecedented actinide carbon triple bonds in uranium methylidyne molecules[J]. Proc Natl Acad Sci U S A, 2007, 104(48): 18919-18924. [11] Andrews L, Wang X, Lindh R, et al. Simple N UF3 and P UF3 molecules with triple bonds to uranium[J]. Angew Chem-Int Edit, 2008, 47(29): 5366-5370. [12] Wang X, Andrews L, Knitter M, et al. Experimental and Theoretical Investigation of Simple Terminal Group 6 Arsenide As MF3 Molecules [J]. J Phys Chem A, 2009, 113(21): 6064-6069. [13] Green DW, Reedy GT. The identification of UN in Ar matrices[J]. J Chem Phys, 1976, 65(7): 2921-2922. [14] Hunt RD, Yustein JT, Andrews L. Matrix infrared spectra of NUN formed by the insertion of uranium atoms into molecular nitrogen[J]. J Chem Phys, 1993, 98(8): 6070-6074. [15] Zhou MF, Andrews L, Li J, Bursten BE. Reaction of laser-ablated uranium atoms with CO: Infrared spectra of the CUO, CUO-, OUCCO, (eta(2)-C-2)UO2, -and U(CO)(x) (x=1-6) molecules in solid neon[J]. J Am Chem Soc, 1999, 121(41): 9712-9721. [16] Zhou MF, Andrews L. Infrared spectra and pseudo- potential calculations for NUO+, NUO, and NThO in solid neon[J]. J Chem Phys, 1999, 111(24): 11044- 11049. [17] Andrews L, Cho HG. Infrared spectrum and structure of CH2=ThH2[J]. J Phys Chem A, 2005, 109(31): 6796-6798. [18] Cho H-G, Andrews L. Methane Activation by Laser- Ablated Th Atoms: Matrix Infrared Spectra and Theoretical Investigations of CH3-Th-H and CH2= ThH2[J]. J Phys Chem A, 2015, 119(11): 2289- 2297. [19] Wang X, Andrews L, Marsden CJ. Infrared spectrum and structure of thorimine (HN=ThH2)[J]. Chem Eur J, 2007, 13(19): 5601-5606. [20] Wang X, Andrews L, Marsden CJ. Reactions of Uranium Atoms with Ammonia: Infrared Spectra and Quasi-Relativistic Calculations of the U: NH3, H2N-UH, and HN=UH2 Complexes[J]. Chem Eur J, 2008, 14(30): 9192-9201. [21] Andrews L, Wang X, Roos BO. AsUF3Molecule with a Weak Triple Bond to Uranium[J]. Inorg Chem, 2009, 48(14): 6594-6598. [22] Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian09 [CP]. Gaussian, Inc: Wallingford CT, 2009. [23] Dolg M, Stoll H, Preuss H. Energy-adjusted ab initio pseudopotentials for the rare earth elements[J]. J Chem Phys, 1989, 90(3): 1730-1734. [24] Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy[J]. Phys Chem Chem Phys, 2005, 7(18): 3297-3305. [25] Weigend F. Accurate Coulomb-fitting basis sets for H to Rn[J]. Phys Chem Chem Phys, 2006, 8(9): 1057- 1065. [26] Bader RFW. Atoms in Molecules: A Quantum Theory [M]. Oxford, UK: Oxford University Press, 1990. [27] Matta CF, Boyd RJ. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design[M]. Weinheim: WILEY-VCH Verlag GmbH & Co KGaA, 2007. [28] George L, Kalume A, Esselman BJ, et al. Spectroscopic and computational studies of matrix-isolated iso-CHBr3: Structure, properties, and photo-chemistry of iso- bromoform[J]. J Chem Phys, 2011, 135(12): 124503. [29] Mayer I, Salvador P. Overlap populations, bond orders and valences for 'fuzzy' atoms[J]. Chem Phys Lett, 2004, 383(3-4): 368-375. [30] Huzinaga S, Miguel B. A comparison of the geometrical sequence formula and the well-tempered formulas for generating GTO basis orbital exponents[J]. Chem Phys Lett, 1990, 175(4): 289-291. [31] Huzinaga S, Klobukowski M. Well-tempered Gaussian basis sets for the calculation of matrix Hartree-Fock wavefunctions[J]. Chem Phys Lett, 1993, 212(3-4): 260-264. [32] Lu T, Chen F. Multiwfn: A multifunctional wave- function analyzer[J]. J Comput Chem, 2012, 33(5): 580-592. [33] Pyykko P, Atsumi M. Molecular Double-Bond Covalent Radii for Elements Li-E112[J]. Chem Eur J, 2009, 15(46), 12770-12779. [34] Espinosa E, Alkorta I, Elguero J, Molins E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X-H center dot center dot center dot F-Y systems[J]. J Chem Phys, 2002, 117(12): 5529-5542. [35] Arnold WD, Oldfield E. The chemical nature of hydrogen bonding in proteins via NMR: J-couplings, chemical shifts, and AIM theory[J]. J Am Chem Soc, 2000, 122(51): 12835-12841. [36] Jenkins S, Morrison I. The chemical character of the intermolecular bonds of seven phases of ice as revealed by ab initio calculation of electron densities[J]. Chem Phys Lett, 2000, 317(1-2): 97-102. [37] Grabowski SJ, Sokalski WA, Leszczynski J. The pos- sible covalent nature of N-H center dot center dot center dot O hydrogen bonds in formamide dimer and related systems: An ab initio study[J]. J Phys Chem A, 2006, 110(14): 4772-4779. Quantum Chemical Studies on the Products of Ce + XF3(X = N, P and As) Reactions LI Yu-qing, GUO Ya-jie, HUANG Zheng-guo (College of Chemistry, Tianjin Normal University, Tianjin 300387, China) The structures, energies and spectra of the possible products (CeXF3, XCeF3, FXCeF2and F2XCeF) of Ce reacting with XF3(X = N, P and As) were studied by ab initio methods. The bonding properties of these molecules were studied by atom-in molecule (AIM), natural population analysis (NPA), Fuzzy bond order (FBO) and Pipek-Mezey localized orbital methods. The results show that all molecules are triplet. For XCeF3, FXCeF2and F2XCeF, the thermodynamic stability of nitride is the highest, followed by arsenide, and that of phosphide is the lowest. However, the case for CeXF3is the opposite. The AIM results indicate that most of Ce-X, X-F and Ce-F bonds show mainly close-shell interaction character and have partial covalent character. Due to the agostic interaction in F2XCeF, the Ce-F1 (or F2) bond shows close-shell interaction character, and the BCP is closed to RCP, which indicates that the stability of the X-Ce-F1 (or F2) ring is weak. electronic structure; ab initio; atom-in-molecule (AIM); agostic effect O641.12 A 1009-9115(2020)03-0029-10 10.3969/j.issn.1009-9115.2020.03.008 天津市自然科学基金项目(18JCYBJC90000) 2019-10-25 2020-04-03 李宇情(1995-),女,天津人,硕士研究生,研究方向为计算化学。 黄正国(1972-),男,山东阳谷人,博士,副教授,硕士生导师,研究方向为计算化学。 (责任编辑、校对:琚行松)
2.2 能量

2.3 AIM分析


2.4 NPA分析


3 结论
