线粒体损伤介导的药源性脏器损伤评价模型研究进展
2020-06-24魏桂林何青青金强杨凌
魏桂林 何青青 金强 杨凌

摘要 线粒体是细胞的能量工厂,同时线粒体还参与细胞分化、维持细胞内环境平衡、细胞信息传递和调节细胞凋亡等过程。线粒体受到核DNA和线粒体DNA的双重调控,使得线粒体对于各种外源及内源性毒性物质更加敏感。药物破坏线粒体结构与功能可导致细胞坏死或凋亡。近年来研究发现多种脏器细胞线粒体是药物毒性作用的主要靶标之一,药物可通过多种途径致线粒体毒性,线粒体损伤是许多药物器官毒性反應早期的重要特征。本文主要综述常见的诱导不同器官线粒体毒性的临床药物并简要探讨药物介导线粒体毒性的机制。此外,我们还将重点讨论目前常用的线粒体毒性评价模型和评价技术,以期为预防和诊断药源性器官损伤提供思路。
关键词 线粒体;线粒体毒性;器官毒性;临床药物;中药;评价模型;评价技术;高通量
Research Progress on Evaluation Model of Drug-induced Organ Damage Mediated by Mitochondrial Damage
WEI Guilin,HE Qingqing,JIN Qiang,YANG Ling
(Institute of Interdisciplinary Integrative Medicine Research,Shanghai University of Traditional Chinese Medicine,Shanghai 201203,China)
Abstract Mitochondria are the energy factories of cells.Meanwhile,mitochondria are also involved in processes such as cell differentiation,maintenance of the intracellular homeostasis,cell information transmission and regulation of cell apoptosis etc.Mitochondria are dually regulated by nuclear DNA and mitochondrial DNA,making mitochondria more sensitive to various exogenous and endogenous toxic substances.The destruction of mitochondrial structure and function by drugs can lead to cell necrosis or apoptosis.In recent years,studies have found that the mitochondria of various organ cells are one of the main targets of drug toxicity.Drugs can cause mitochondrial toxicity through a variety of ways.Mitochondrial damage is an important feature of many drug organ toxicity in the early stage.This paper mainly reviews common clinical drugs that induce mitochondrial toxicity in different organs and briefly discusses the mechanism of drug-mediated mitochondrial toxicity.In addition,we will also focus on the current commonly used mitochondrial toxicity evaluation models and evaluation techniques in order to provide ideas for the prevention and diagnosis of drug-mediated organ damage.
Keywords Mitochondria; Mitochondrial toxicity; Organ toxicity; Clinical drugs; Chinese medicines; Evaluation model; Evaluation technology; High throughput
中图分类号:R285.5文献标识码:Adoi:10.3969/j.issn.1673-7202.2020.23.001
线粒体是由双层膜包裹的细胞器,是细胞的主要能量来源,线粒体以ATP的形式储存能量,并以ATP水解释放能量以供生命活动之需,如用于肌肉收缩和细胞分裂,蛋白质生物合成、折叠和降解以及膜电位的产生和维持等[1-2]。除了细胞呼吸和产生能量外,线粒体还参与其他代谢过程,包括糖类、脂肪以及蛋白质三大营养物质的最终氧化,氨基酸、血红素的生物合成,钙信号传导、应激反应以及一般的细胞信号中枢。此外,线粒体还参与细胞凋亡、坏死与衰老等过程。因此,线粒体在人类身体健康中发挥着重要作用。
线粒体功能紊乱通常会诱发细胞损伤乃至死亡,故而线粒体也是许多疾病治疗的靶点[3]。开发靶向干扰线粒体功能的药物一直也是很多医药公司设计和开发针对治疗目的的主要策略[4]。但是,对于大多数制药公司而言,药物的安全性愈发变的不可忽视。越来越多的实验和临床研究表明,线粒体是许多药物毒性产生的靶点,严重不良反应可能是药物或代谢中间体直接或间接破坏线粒体结构与功能造成的[5-6]。扰乱线粒体的药物通常是通过抑制电子传递链的呼吸复合物,抑制或解偶联氧化磷酸化,诱导线粒体氧化应激,降低线粒体膜电位,扰乱细胞内钙离子稳态,或抑制DNA复制、转录或翻译等过程产生的[7]。线粒体毒性是上市药物撤药以及器官毒性的一个重要原因,FDA等机构也明确要求线粒体毒性作用应纳入药物的安全评价中[8]。因此,在药物发现的早期阶段检测药物诱导的线粒体功能障碍是至关重要的,构建高效地线粒体毒性评价模型来减少候选药物的后期消耗,降低药物不良反应,并设计出更安全的药物对于整体医药产业、人类健康意义重大。
本综述主要围绕药物诱导器官、组织细胞的线粒体毒性,并举例说明了药物诱导的线粒体毒性的多种机制;概述了药物诱导的线粒体功能改变的传统和新颖评价模型和评价技术,以期为预防、诊断、干预或改善外源性药物引起的脏器损伤和相关疾病提供思路。
1 线粒体结构与功能
线粒体由双层膜结构将线粒体分为外膜、膜间隙、内膜和线粒体基质四大部分。外膜平整光滑,相当于细胞膜,外膜主要参与调控线粒体与内质网等细胞器以及细胞质之间的物质交换。外膜和内膜之间的区域是膜间隙,由于外膜上分布有孔蛋白构成的通道,通透性很高,所以膜间隙中的离子环境与胞质的几乎相同。内膜包裹着线粒体基质,蛋白含量高,部分内膜向内折叠形成嵴,在不同种类的细胞中,线粒体嵴的数目、形态和排列方式有较大差别,嵴的形成增加了线粒体内膜的表面积,因此,内膜通透性低并且在内膜上进行着更多的生化反应。线粒体基质含有参与三羧酸循环、脂肪酸氧化、氨基酸降解等生化反应的酶等众多蛋白质,所以较细胞质基质黏稠,线粒体基质中一般还含有线粒体DNA,RNA和核糖体。
线粒体是细胞的动力工厂,是真核生物进行氧化代谢的部位,是糖类、脂肪和氨基酸最终氧化释放能量的场所,细胞中95%以上的能量来源于线粒体[9]。同时,线粒体还参与调解钙离子稳态,活性氧的产生以及细胞内信息传导平台,调控细胞死亡,如凋亡、坏死以及自噬等。
2 线粒体损伤模式及机制
线粒体损伤包括结构和功能上损伤。很多药物可以破坏线粒体膜完整性,导致线粒体裂解,从而丧失功能[10-11]。另一方面,绝大部分线粒体毒性药物都是通过破坏线粒体功能从而诱发线粒体能量障碍,最后引起细胞的凋亡或坏死。药物引起线粒体功能的方式主要包括4个方面:1)产生过量ROS;2)钙离子紊乱和线粒体通透性转变;3)阻断线粒体呼吸链;4)损伤线粒体DNA[12-14]。见图1。
线粒体电子呼吸链泄露的电子攻击O2是线粒体产生ROS的主要原因,这些ROS会攻击磷脂,蛋白质和线粒体DNA从而损伤线粒体功能[15]。正常情况下,线粒体超氧化物歧化酶将超氧化物分解为过氧化氢,然后通过谷胱甘肽过氧化物酶将其转化为水。此外,胞质超氧化物歧化酶和过氧化氢酶可提供细胞具有针对ROS的其他抗氧化剂防御机制。但是,很多外源性药物或代谢生成的活性中间体可直接产生大量的ROS,引起氧化应激[16],线粒体作为产生ROS的主要细胞器,因此也成为主要被攻击
研究表明,药物或其反应性代谢产物可通过改变线粒体的结构或功能来诱导线粒体通透性转换孔(Mitochondrial Permeability Transition,mPTP)。目前认为主要有2种机制,一种机制涉及促凋亡分子BAX/BAK在线粒体外膜上形成孔[19]。由药物或其他有毒化学物诱导的细胞应激与BH3蛋白的激活有关。这些蛋白质通过激活BAX/BAK促进线粒体介导的细胞死亡,进而诱导线粒体外膜上的孔形成。通过这些孔释放的线粒体促凋亡蛋白CytC,EndoG和AIF启动细胞凋亡。第二种机制是由于钙稳态的改变或mtROS产生的增加而引起的mPTP诱导,从而导致线粒体内膜破裂[20]。线粒体是钙离子储存的主要场所之一,在线粒体内,钙离子会增加三羧酸循环和ATP产生至关重要的几种酶的活性,激活了线粒体的代谢,增加了有氧条件下许多能量消耗过程中ATP的供应。线粒体对钙离子的摄取和外排共同维持细胞内钙离子的稳态,并与内质网、细胞膜共同参与调节胞质内游离钙离子浓度,维持细胞内钙离子的稳态。很多药物或代谢物会直接或间接破坏钙离子平衡,导致大量线粒体钙离子外流进入胞质中,钙离子浓度的剧降抑制ATP合成,最终引起氧化磷酸化过程障碍。
化合物在线粒体中氧化产生的NADH和FADH2通过电子传递链(Electron Transfer Chain,ETC)将质子和电子传给O2生成水分子,电子传递链的抑制在某种程度上意味着细胞能量代谢障碍,甚至细胞生命的终结。许多药物或其亲电子代谢物可以抑制线粒体电子传输链并导致活性氧(ROS)形成,从而触发MMP和细胞色素c的释放[21]。另一方面,在完整的线粒体内,电子传递与磷酸化是紧密偶联在一起的,很多药物可通过氢穿梭穿过膜或通过通道形成或仅通过破坏脂质双层来充当解偶联剂,导致电子传递与ATP的形成这2个过程分开,则只进行电子传递而不能生成ATP[22]。
线粒体DNA是独立于细胞核染色体外的基因组,能够单独进行复制、转录及合成蛋白质。但是由于mtDNA是裸露的,缺乏保护,且处于线粒体呼吸链产生的高活性氧环境中,因此,mtDNA极易受氧自由基的侵害[16]。并且氧化损伤引起的线粒体DNA突变或其基因产物丢失又会进一步促进活性氧的释放,进而又加重线粒体DNA的损伤[23]。
3 药源性线粒体毒性
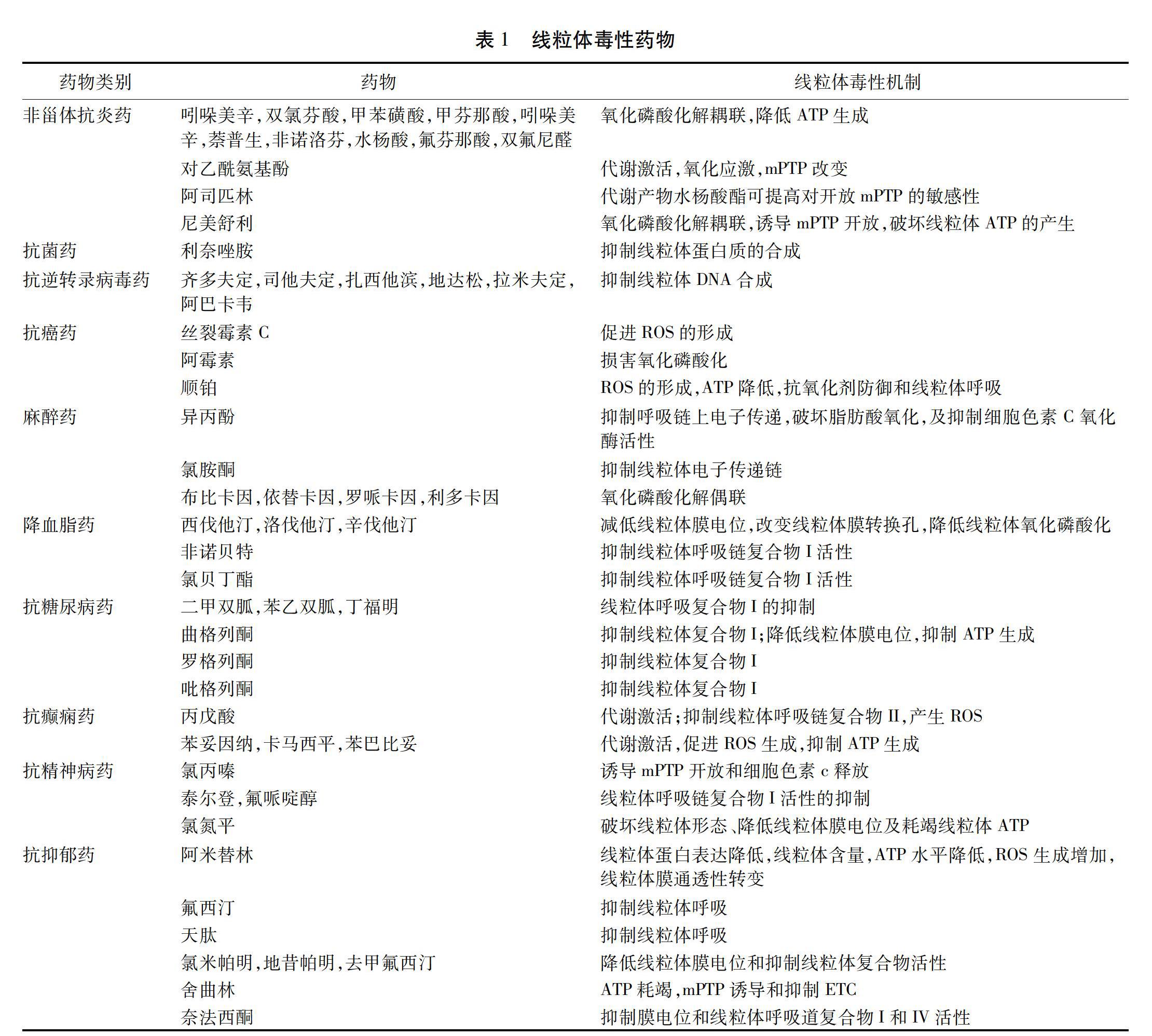
线粒体由于其膜腔呈弱碱性(pH≈8),且处于负膜电位状态,使得很多带正离子药物很容易由于电势差而进入线粒体内部,大量富集造成线粒体功能障碍。不同类别药物由于其各自独特的结构特征,使其对线粒体毒性机制存在差异。以下列举了引起线粒体不同毒性机制的药物。见表1。
3.1 非甾体抗炎药 非甾体抗炎药(NSAIDs)是一类不含有甾体结构特征的药物,这类药物通常需要弱酸性基团(羧酸)结合到COX2酶的花生四烯酸盐结合位点上,抑制COX2活性来发挥药效,具有抗炎、抗风湿、止痛、退热和抗凝血等作用。越来越多证据表明该酸基团的脂溶性与细胞线粒体内膜磷脂相互作用,起质子体的作用,损伤线粒体功能。吲哚美辛,双氯芬酸和其他NSAIDs,包括选择性COX-2抑制剂,在离体大鼠心脏制剂中显示出可解除呼吸耦合并减少ATP产生[22]。另外,在大鼠心肌细胞和鼠新生心肌细胞中,NSAIDs可能通过抑制线粒体电子运输链来增加ROS的产生。Masubuchi等人發现大多数NSAID的结构成分二苯胺可解偶联氧化磷酸化,降低肝脏ATP含量,导致线粒体肿胀,从而诱发大鼠肝制备中的肝细胞损伤[24]。NSAIDs解耦效果的顺序为氟芬那酸>双氟尼醛>甲苯磺酸>甲芬那酸酸>双氯芬酸>吲哚美辛>萘普生,非诺洛芬>水杨酸[25]。而不含二苯胺结构的对乙酰氨基酚和阿司匹林也具有明显的肝细胞线粒体毒性,则是因为对乙酰氨基酚可以被代谢激活,其代谢物与线粒体蛋白结合并诱导氧化应激,导致线粒体通透性转变[26]。同样,阿司匹林的主要代谢产物水杨酸酯可提高对开放mPTP的敏感性。尼美舒利可通过解耦线粒体呼吸并诱导离体大鼠肝细胞和培养的人肝癌细胞中的mPTP来破坏线粒体ATP的产生,表明该药物对线粒体具有直接毒性,而不需要代谢激活[27]。
3.2 抗菌药 抗菌药根据结构可分为喹诺酮类药物、β-内酰胺类、大环内酯类和氨基糖苷类。喹诺酮类,氨基糖苷类和β-内酰胺类被证明可通过抑制线粒体ETC导致线粒体功能受损,从而导致哺乳动物细胞中ROS的生成增加,进而导致氧化性组织损伤[28]。体外研究表明氨基糖苷类可以靶向线粒体核糖体,干扰蛋白质合成,β-内酰胺类抑制线粒体肉碱/酰基肉碱转运蛋白而抑制脂肪酸β氧化,严重时导致肾毒性[29]。相较而言,四环素和大环内酯类抗菌药似乎对线粒体功能没有不利影响。由于细菌和线粒体核糖体的相似性,用于严重耐药革兰氏阳性细菌感染的利奈唑胺,可抑制线粒体蛋白质的合成,特别是抑制呼吸链复合体IV[30]。
3.3 抗逆转录病毒药物 抗逆转录病毒药物主要通过抑制線粒体DNA合成和氧化损伤DNA。尽管抗逆转录病毒药物结构差异导致不同程度线粒体功能异常,总体上这类药物通过抑制线粒体DNApoly导致mtDNA缺失,从而损害线粒体呼吸功能,引起细胞内脂质蓄积和高乳酸血症[31]。齐多夫定(AZT)或司他夫定(d4T)作为胸腺嘧啶类似物;去羟肌苷(腺苷类似物);扎西他滨或拉米夫定为胞嘧啶类似物;阿巴卡韦和阿巴卡韦(鸟嘌呤类似物)是核苷和核苷酸逆转录酶抑制剂,用于预防AIDS患者中HIV病毒的复制。然而,由于这些药物也抑制线粒体DNA聚合酶γ,因此在患病患者中线粒体DNA复制也受到部分影响。这些药物线粒体毒性大小依次为:扎西他滨>地达松>司他夫定>拉米夫定>齐多夫定>阿巴卡韦[31]。
3.4 抗癌药 抗癌药物依据作用机制不同一般分为抑制DNA合成、直接破坏DNA结构或与DNA结合、抑制蛋白质的合成、抑制有丝分裂等四大类。研究显示醌类抗肿瘤药物如丝裂霉素C、阿霉素等具有显著线粒体毒性。丝裂霉素C的对苯二酚结构可作为亲电试剂与基因组DNA以及单和二烷基化DNA形成交联,从而防止肿瘤细胞增殖。但是,在这些有氧条件下,丝裂霉素C引起线粒体功能障碍,因为线粒体还原酶代谢产生的丝裂霉素半醌自由基中间体可与氧反应形成氧化还原循环,从而形成可氧化生物分子的ROS。抗肿瘤药物阿霉素介导的线粒体功能障碍似乎是由线粒体外膜NADH:b5还原酶和内膜复合物I还原而形成的半醌自由基间接引起的,它通过使电子从呼吸链转移而损害了氧化磷酸化。随后,半醌中间体与氧气反应形成超氧化物,变成过氧化氢[32]。顺铂是另一种用于实体瘤的抗癌药物,顺铂诱导线粒体毒性涉及ROS的形成,ATP降低,抗氧化剂防御和线粒体呼吸[33]。
3.5 麻醉药 体外研究表明,全身麻醉药主要通过抑制电子传递链来降低线粒体功能。在短期全身麻醉药(挥发性麻醉剂,异丙酚,神经肌肉阻滞剂,苯二氮艹卓类药物和阿片类药物)抑制线粒体呼吸而不影响线粒体复合物的酶活性或氧化磷酸化偶联[34]。麻醉药异丙酚线粒体毒性可能包括抑制呼吸链上电子传递,破坏脂肪酸氧化,及抑制细胞色素C氧化酶活性[35]。在一项研究中,异丙酚通过消耗完仔猪大脑中的糖原和线粒体ATP来改变线粒体的代谢。氯胺酮的超临床浓度可通过线粒体途径诱导神经元凋亡,涉及线粒体膜电位降低,ROS水平升高,线粒体裂变增加和Caspase-3活性[36]。而氯胺酮增加ROS,降低线粒体ATP的内在机制主要是由于其可抑制线粒体电子传递链[37]。局部麻醉药布比卡因和依替卡因的严重心脏毒性和肌毒性限制了它们的用途,并归因于其线粒体的解偶联能力。作为高度亲脂性的两亲性胺,它们可以通过质子传递机制,通过质子穿过线粒体膜并刺激分离的线粒体呼吸[38]。布比卡因还比罗哌卡因更有效地使心脏细胞线粒体解偶联,特别是在复合物I处,而利多卡因在解偶联上的活性低得多[39]。布比卡因诱导的肌毒性和肌病的机制被认为是由于线粒体毒性导致ATP耗竭,Ca2+超载和ROS形成,从而导致通透性转变开孔[40]。
3.6 降血脂药 他汀类药物是抑制HMG-CoA还原酶抑制药,是目前最有效的降脂药物。长期使用他汀药物可导致肝毒性和肌毒性,研究显示这些不良反应与线粒体功能紊乱有关。西伐他汀,洛伐他汀,辛伐他汀可以显著降低线粒体膜电位,改变线粒体膜转换孔,降低线粒体氧化磷酸化从而破坏肝细胞线粒体功能[41]。而他汀类药物骨骼肌线粒体毒性则主要与消耗线粒体mtDNA关联[42]。另一类降脂药如非诺贝特、氯贝丁酯也有一定的线粒体毒性,它们则主要通过抑制线粒体呼吸链复合物I活性而导致线粒体功能障碍[43]。
3.7 抗糖尿病药 双胍类药物(二甲双胍、苯乙双胍、丁福明)的抗糖尿病作用以及诱导的乳酸性酸中毒都可能归因于线粒体呼吸复合物I的抑制[44]。复合体I的抑制机制尚不清楚,但可能涉及双胍分子的烃部分与膜磷脂的烃链结合,质子化胍基的正电荷与磷脂磷酸基相互作用。噻唑烷二酮类降糖药包括曲格列酮,罗格列酮和吡格列酮,这些药也通过抑制线粒体复合物I,从而降低糖异生作用,并可能具有抗糖尿病作用[45]。它们通过抑制复合物I活性,引起骨骼肌或大鼠肝匀浆中的乳酸释放,从而导致毒性。曲格列酮可通过降低线粒体膜电位,减少ATP合成而诱导线粒体毒性。
3.8 抗癫痫药 研究显示许多抗癫痫药具有不同程度的线粒体毒性。丙戊酸是一种广谱抗癫痫药,用量不适时也会引起线粒体毒性,其中肝毒性最明显。丙戊酸可在肝脏中被代谢激活,活性产物可结合到还原的乙酰辅酶A上,从而被摄入线粒体基质内,抑制多种线粒体酶并降低线粒体脂肪酸氧化[46]。此外,研究还发现丙戊酸可抑制线粒体呼吸链复合物II,诱导脂质过氧化和膜电位降低,引起ROS产生增加,进而引起线粒体功能异常[47]。其他抗癫痫药如苯妥因纳、卡马西平,苯巴比妥也可诱发线粒体功能障碍,它们对线粒体毒性大小依次为苯妥因纳>苯巴比妥>卡马西平[48]。
3.9 抗精神病药 抗精神病药依据结构主要包括吩噻嗪类(氯丙嗪)、硫杂蒽类(泰尔登)、丁酰苯胺(氟哌啶醇)及其他类药物。研究表明,很多抗精神病药会破坏线粒体功能,特别是对线粒体呼吸链复合物I活性有抑制作用。研究抗精神病药(典型的抗精神病药)对人脑微血管内皮细胞的作用,证实了这些发现,其中线粒体呼吸链复合物I和III的活性被所有抗精神病药抑制[48]。吩噻嗪类如氯丙嗪等衍生物可通过mPTP开放和细胞色素c释放导致线粒体损伤,最后引起细胞凋亡[49]。氯氮平的线粒体毒性机制则主要包括破坏线粒体形态、降低线粒体膜电位及耗竭线粒体ATP[50]。
3.10 抗抑郁药 大量的体外和体内研究表明,常用的抗抑郁药会抑制线粒体功能并增加氧化应激。阿米替林诱导的线粒体毒性与线粒体蛋白表达降低,线粒体含量,ATP水平降低,ROS生成增加,线粒体膜通透性转变有关[51]。其他抗抑郁药,例如氯米帕明,地昔帕明,天肽,氟西汀对线粒体有作用。研究显示氟西汀和天肽可有效抑制线粒体呼吸,但氯米帕明,地昔帕明和去甲氟西汀则由于降低线粒体膜电位和抑制线粒体复合物活性而导致凋亡[52]。长时间使用舍曲林导致ATP耗竭,mPTP诱导和抑制ETC而导致不可逆的线粒体损伤和细胞死亡[53]。奈法西酮由于抑制膜电位和线粒体呼吸道复合物I和IV活性,从而引起严重肝毒性[54]。
4 線粒体毒性与脏器损伤
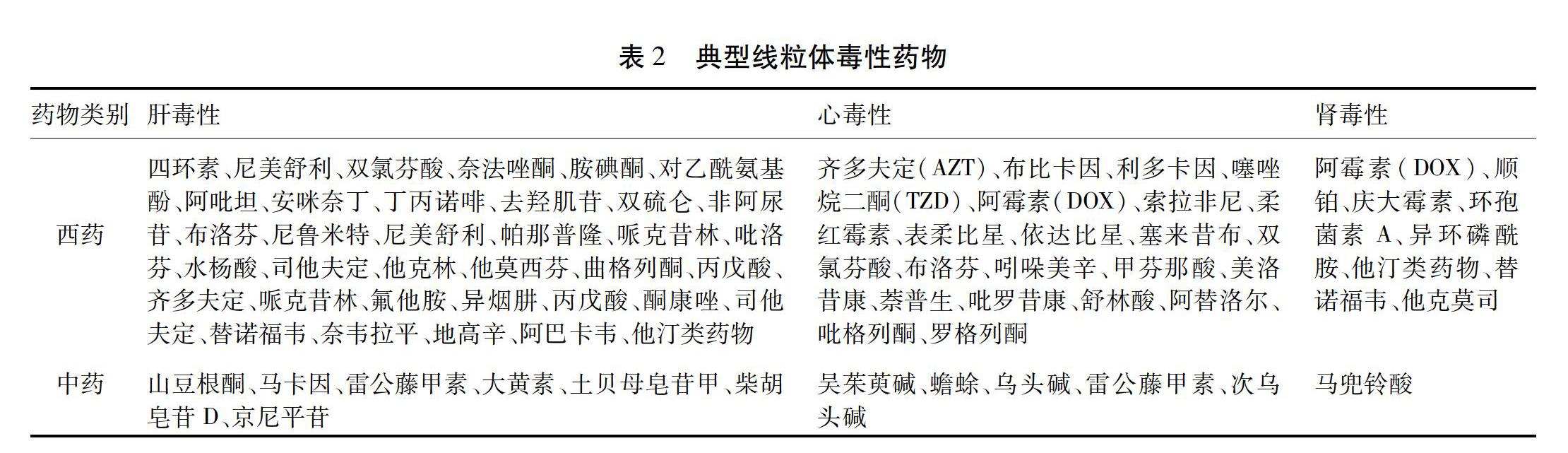
每年都有相当大比例的药物由于出现临床前期或中期未发现的不良反应而被撤出市场。越来越多的证据表明,这些不良反应很多是线粒体功能障碍引起的心脏、肝脏和肾脏毒性作用。下表列举了主要经由线粒体途径的心肝肾毒性药物[3,13,55-56]。见表2。
4.1 线粒体损伤致肝毒性 肝脏作为药物代谢的重要脏器,也是药物引发损伤的主要靶器官。一些抗病毒药物、抗肿瘤药物和抗生素等可显著诱导肝脏线粒体损伤。很多肝毒性药物主要通过改变线粒体结构、酶的活性或减少线粒体DNA的合成,进一步破坏脂肪酸β氧化和肝细胞的氧化能力,最终诱发肝损伤。线粒体在哺乳动物的肝细胞中含量非常丰富,其参与肝细胞内电解质稳态平衡的调控、离子跨膜转运、氧自由基生成、细胞信号转导和细胞凋亡等一系列的细胞生理活动。同时,线粒体也是最容易受损的一个细胞器,其可以显示细胞受损的程度。越来越多的研究表明,线粒体损伤可能是药源性肝损伤诱发因素和经由途径[13]。因此,认识和深入研究线粒体损伤在药源性肝损伤发生中的重要作用,可为药源性肝损伤临床早期诊断、预防及治疗提供新思路。药物诱导的线粒体损伤主要包括结构改变和功能紊乱。线粒体结构的改变主要表现为线粒体肿胀、体积显著增大、内外膜完整性破坏甚至出现膜溶解、线粒体脊断裂及大量空泡化,线粒体结构基本丧失。药物经由线粒体诱发肝损伤的机制主要有干扰线粒体呼吸链上的电子传递、氧化磷酸化的解偶联、抑制线粒体脂肪酸β氧化、干扰线粒体Ca2+紊乱、促进mPTP开放、破坏线粒体DNA等途径。胺碘酮是常用抗心律失常药物,研究显示胺碘酮诱导肝细胞线粒体功能障碍而发生肝损伤的主要机制是其抑制呼吸链复合物Ⅰ,阻断呼吸链中电子的传递,最终降低ATP的生成[17]。中草药雷公藤对细胞免疫和体液免疫均有抑制作用,其主要的有效成分和毒性成分为雷公藤甲素。雷公藤甲素引起肝损伤与直接损害肝线粒体的结构和功能有关,线粒体损伤可能是雷公藤甲素致肝损伤的机制之一[57]。研究表明雷公藤甲素肝毒性主要体现在肝细胞线粒体呼吸链的电子传递障碍,线粒体膜电位降低,显著地抑制线粒体呼吸链复合物Ⅰ、Ⅳ等。二苯胺类非甾体抗炎药不仅可以降低线粒体膜电位,还可刺激基础氧消耗并抑制ADP呼吸,使得线粒体氧化磷酸化解耦联,ATP生成急剧减少,导致能量代谢障碍从而诱发肝损伤。丙戊酸是一种抗惊厥药,越来越多研究表明丙戊酸可以消耗肉碱,减少辅酶A供应和抑制β氧化的三大功能酶,从而阻碍线粒体β氧化,并且其代谢产物还能与脂质竞争相关的酶,这些都是丙戊酸诱导肝毒性的主要原因[47]。一些抗结核药如异烟肼和利福平的肝毒性则主要与线粒体氧化应激和线粒体mPTP异常开放密切相关[58]。此外线粒体具有自己独立的遗传信息,能够独立进行基因的转录、翻译及蛋白质的表达。很多核苷类及单核苷酸类逆转录酶抑制剂在临床长期治疗时,会抑制线粒体DNA聚合酶,导致线粒体酶合成受阻、特别是氧化磷酸化中ETC关键蛋白的表达受到影响,导致严重的能量代谢障碍。并且这些药物也还能与DNA聚合酶结合,终止DNA复制,从而减少mtDNA含量而损伤线粒体[59]。
4.2 线粒体损伤致肾毒性 肾脏由于其功能和解剖学特点,是药物毒性的重要靶器官之一,药物诱导的急性肾损伤是药物研发过程中常见的问题。急性肾毒性以近端肾小管损伤为主要特征,表现为肾小管肿胀、肾小管上皮细胞坏死、管型形成、间质炎性反应及细胞凋亡等。肾小管,尤其是近端小管,对离子、葡萄糖及营养物质的重吸收均需要通道和转运蛋白的主动运输才能完成,需要维持线粒体正常功能以保证能量供应[55]。肾脏是一个高能量需求的器官。由于肾脏的再吸收过程对能量(ATP)的持续和高度依赖,使得线粒体功能障碍成为可能参与药物诱导的急性肾毒性的关键发病机制。因此,药物引起的线粒体损伤可以中断化学物质的再吸收过程,在急性肾毒性的发病机制中起着至关重要的作用。常见的具有引起急性肾毒性倾向的药物有抗生素、化疗药物、免疫抑制药物、抗病毒药物、中药、非甾体抗炎药物、质子泵抑制剂等。诸多研究均在AKI模型中观察到肾小管上皮细胞线粒体嵴消失、线粒体碎片化及质量下降、线粒体肿胀等表现。
近端肾小管对许多化学物质的重吸收过程严重依赖于Na+/K+ATP酶产生的电化学梯度。Na+/K+ATP酶是一种存在于多种细胞质膜中的酶。这种泵在不同的生理过程中起着至关重要的作用,如从胃肠道吸收化学物质或通过神经元传递脉冲。Na+/K+ATP酶将Na+泵出细胞,同时将K+泵入细胞,两者都是逆浓度梯度转运。Na+/K+ATP酶活性依赖于ATP。在肾脏中,Na+泵出提供的驱动力用于将几种化学物质,如氨基酸、葡萄糖、磷酸盐、蛋白质、维生素和其他一些有机化合物导入近端小管细胞,进而进入血流。因此,细胞内ATP含量的任何改变都会损害其功能。肾小管内层细胞含有大量线粒体,为化学物质的主动转运和再吸收提供足够的能量。近端小管细胞具有较高的代谢率,可通过线粒体氧化磷酸化产生大量ATP。近端小管细胞对ATP的高度依赖性使其成为外源性毒物诱导的线粒体损伤和能量危机的潜在靶细胞。对线粒体功能的干扰会破坏能量代谢和ATP的产生。因此,细胞ATP耗竭可导致Na+/K+ATP酶抑制和破坏化学物质再吸收过程和血清电解质的紊乱。
有毒物质靶向蓄积于肾脏也是药物诱导肾脏毒性的一个主要原因,这与肾小管细胞膜表面蛋白、有机阳离子转运体、铜离子转运体等密切相关。由于近端小管细胞线粒体丰富,因此药物在近端小管的蓄积,势必会损伤其内含量丰富的线粒体,进而触发线粒体损伤。研究表明线粒体参与了药物诱导的急性肾损伤的病理过程。线粒体动力学失衡将导致线粒体碎片化,进而可导致线粒体质量下降[60]。线粒体碎片化是AKI发生时线粒体最早期变化,发生于肾损伤及细胞凋亡前。线粒体动力学紊乱一方面参与肾小管上皮细胞损伤机制,由此参与多种因素导致的肾小管损伤。肾小管细胞凋亡和氧化应激损伤是药物诱导肾毒性的明显病理学特征,其中,线粒体介导的内源性细胞凋亡途径对于药物诱导的肾毒性的发生发展十分关键。同时由于肾脏近端小管的抗氧化防御系统相对薄弱,而线粒体又是机体活性氧生成的主要场所,因而肾脏对线粒体功能障碍更加敏感。另一方面,肾近端小管上皮细胞由于对这些药物的高吸收和积累能力而经常受到药物的影响。这一事件将增加由于过度暴露于外源性药物引起的肾脏组织损伤的风险。综上所述,药物在近端小管的蓄积是DAKI的物质基础,结合肾脏近端小管线粒体富集这一生理特点以及相关病理学特征,可认为线粒体损伤是药源性急性肾损伤的重要病理学机制。
总而言之,D-AKI过程中线粒体损伤主要表现为结构改变与功能障碍,具体涉及到氧化应激损伤、线粒体动力学过程紊乱、线粒体自噬水平不足、线粒体生物合成不足、线粒体呼吸链缺陷等多方面因素。多种因素彼此之间相互交叉、相互关联,共同加剧了D-AKI的发生发展。
4.3 线粒体损伤致心毒性 药物心脏毒性是因外源性药物对心血管系统造成多种复杂的病理生理损害,临床表现为心肌炎、心肌病、心律失常、心瓣膜损害、心肌缺血及与心肌梗死等一系列心脏功能和器质性的改变。心脏结构复杂,有毒药物可通过与各种受体、离子通道的相互作用,参与氧化应激反应,造成细胞器损伤及影响细胞凋亡等多种途径对其造成损伤。线粒体在心肌细胞中占了很大的比例,位于肌原纤维之间、内质膜下方。线粒体显著的地位和数量保证了为心脏收缩、新陈代谢和离子动态平衡提供集中高效的ATP运输系统[61]。线粒体在心肌细胞的存亡中起着关键的作用。多种证据表明,多种常用的抗癌药,抗病毒化合物和抗糖尿病或非法滥用药物,例如乙醇,可卡因,甲基苯丙胺,摇头丸和合成大麻素涉及与线粒体相关的直接或间接毒性,主要包括干扰线粒体呼吸链(例如,解偶联)或重要线粒体酶的抑制(氧化磷酸化,三羧酸循环,线粒体DNA复制,ADP/ATP易位),最终导致线粒体膜电位丧失,线粒体氧化/硝化应激增加,和细胞死亡。需要更透彻的了解线粒体心血管毒性的常见机制,以开发灵敏且高通量的线粒体毒性筛选方法和体内模型,以更好地预测新型化合物的不可预见的心脏毒性问题,并基于更具选择性的靶向性设计新的心脏保护策略预防上述普通药物严重心血管不良后果的特定线粒体过程。
5 线粒体毒性评价模型
近年来,越来越多的研究表明,线粒体是药物毒性作用的重要靶标,其功能异常通常会导致或加速细胞死亡,线粒体毒性评价已成为候选药物安全性评价的重要指标[62]。因此评估线粒体的各项指标对于内源性或外源性物质的安全性评价意义重大。
合适的实验模型对于线粒体毒性的评估至关重要。尽管体内啮齿动物实验提供了相当多的毒性数据,但啮齿动物模型存在种属差异大,通量低,毒性评价周期长等问题。离体的线粒体或培养的细胞体外毒性实验被广泛的用于线粒体毒性机制评价,不但可以进行高通量的体外筛选,而且通常比啮齿动物体内进行的分析更有效。目前基于细胞的线粒体机制效应或毒性模型是不够的,因为培养的细胞系存在高度糖酵解、有氧代谢最小化以及线粒体生理功能的改变等问题。因此,近年来,像斑马鱼、秀丽隐线虫等可进行高通量筛选的体内动物模型逐渐被广泛应用于体内毒性评价[63]。
5.1 离体线粒体 离体线粒体通常被用于体外评估外源性药物对线粒体功能的影响,以确定线粒体毒性在特定器官损伤中的作用。线粒体可以从不同脏器组织、培养的细胞中分离出来。所选组织被机械均质化破坏形成组织匀浆,然后用差速离心法分离出线粒体[64];线粒体也可通过细胞裂解分离,然后进行差速离心分离和纯化[65]。使用离体的线粒体来测量有毒物质对氧气消耗的影响,可以避免细胞或整个生物体中存在任何非线粒体呼吸源,包括过氧化物酶体的呼吸[66]。离体线粒体中的氧化磷酸化作用不受代谢和细胞信号传导的影响,因此,检测离体线粒体中的氧化磷酸化作用只能获得完整细胞或动物整体呼吸作用的一部分信息。但是,线粒體的分离会破坏线粒体结构并可能改变线粒体功能。离体的线粒体类似于球形单位,具有相对均匀的大小和形状,线粒体分离会破坏通常情况下线粒体不均匀的分支结构,并产生相对均匀的球形细胞器[67]。在组织均质化过程中,线粒体网状膜结构会被破裂,必须迅速重新组装才能产生明显的“完整”细胞器。可能在线粒体膜短暂破裂、又重新组装的过程中,可溶性基质酶从线粒体基质中丢失,从而稀释氧化磷酸化或活性氧解毒所需的基质成分,破坏了线粒体内膜及其呼吸链复合物的结构完整性。因此,使用离体的线粒体检测线粒体功能具有一点的缺陷性。
5.2 活细胞水平 目前,药物毒性评价常用的体外细胞模型有正常细胞系、癌细胞系和原代细胞系等,主要采用2D培养的方式,但是2D培养难以维持细胞立体结构形态和细胞间的联系,导致细胞培养与体内器官真实情况相差较大。3D培养跨越二维细胞培养和机体整体,模拟体内环境,3D模型可增强细胞之间的黏附以及维持细胞骨架,增加细胞间交流[68]。细胞形态和信号转导通常比常规的2D细胞培养更具有生理学特性。在平面2D组织培养基质上生长的细胞在形态,细胞-细胞和细胞-基质相互作用以及与在更接近生理环境的3D培养中生长的细胞在功能检测等方面可能存在很大的差异。但3D培养模型模仿体内组织状况的能力各不相同而且缺乏血管系统以及小分子信号传导等,通常模仿静态或者短期状态很难代表体内系统[69]。
原代细胞系是评估外源性药物或内源性物质线粒体毒性的理想模型。然而,原代细胞培养费时,价格昂贵,以及它们在培养中的短暂存活力,共同限制了原代细胞在药物开发领域的效用。由于这些原因,肿瘤来源的、不朽的细胞系已成为用于药物发现和检测线粒体毒性的常用细胞模型。癌细胞具有永生化的分裂能力,而且生长能力比较强。即使在氧气充足的条件下,癌细胞也能通过糖酵解产生几乎所有的ATP,然后进行乳酸发酵[70]。因此,以葡萄糖为底物时癌细胞对线粒体毒物的敏感性较低。细胞有氧呼吸被高浓度的葡萄糖抑制,从早期的细胞培养开始,为了方便,细胞一直在含有25 mm葡萄糖的培养基中生长,这比正常水平高出5倍。在这种情况下,细胞不会大量使用线粒体来产生ATP,因此当评估候选药物在这些细胞中的潜在毒性时,即使存在非常强的线粒体毒素,也不会检测到细胞死亡。一种可能的办法是更换底物,以半乳糖代替培养基中的葡萄糖。半乳糖很难被转换为葡萄糖,无法通过糖酵解产生ATP,用于生长的能量通过谷氨酰胺代谢获得[70-71]。谷氨酰胺被线粒体谷氨酰胺酶分解为谷氨酸,然后通过谷氨酸脱氢酶转化为酮戊二酸,并在三羧酸循环中进一步处理,最后结合磷酸化产生ATP[72-73]。因此,在培养基中用半乳糖代替葡萄糖可迫使细胞依赖氧化磷酸化(OXPHOS)来产生必要的ATP,增加细胞对线粒体毒性物质的敏感性[74-75]。
5.3 体内评估体系 离体的线粒体,培养的细胞和整个生物体之间有许多重要的线粒体功能不同。这些差异包括细胞内能量转移、细胞间基质作用以及核受体等机体的调控作用,可能影响药物对线粒体毒性的敏感性。体内评估体系是在整个生物体的背景下检测线粒体,使其在生理上更具相关性。体内模型也是研究线粒体功能失调对早期发育影响的理想方法[76]。
斑马鱼和秀丽隐杆线虫的体积小,可以像细胞一样培养在多孔板中,繁殖力强且发育迅速,因此非常适合进行体内毒理学和药物发现研究。斑马鱼和秀丽隐杆线虫作为廉价的动物模型,在获得体内关键数据的同时,可测量体内多个功能指标,实现实时高通量药物的体内检测,不仅可以在体内筛选化学药物,而且也可以对其作用机制进行深入分析。也可以进行跨代研究,因为它们繁殖力强而且胚胎是半透明的,可以可视化体内的细胞和亚细胞现象,研究人员可以轻松地监视器官的发育[77-78]。此外,秀丽隐杆线虫和斑马鱼的基因可进行处理,转基因体和遗传突变体也很容易獲得,因此增加了实验可行性。已被用作多个线粒体高通量检测平台的模型,包括使用荧光报告基因分析斑马鱼体内神经元线粒体,利用XFe24细胞能量分析仪测定斑马鱼幼虫线粒体生物能,利用秀丽隐杆线虫检测环境污染物的线粒体毒性等[79-80]。
不同动物、不同组织线粒体对毒性物质的敏感性不同[81-83]。有文献报道,在相同浓度的胆红素暴露下,脑线粒体磷酸化几乎完全解耦联,而肝脏线粒体仅不到50%解耦联。线粒体产能的阈值会随着年龄的增长或缺乏运动而减少,这表明,与年轻活跃的啮齿动物比较,久坐不动的年老啮齿动物可能更适合研究药物诱发的线粒体功能障碍[84-85]。因此,动物模型的选择应考虑线粒体的易感性。
6 线粒体毒性评价技术
线粒体结构复杂,功能多样,不仅是细胞的供能场所,还参与细胞死亡、肿瘤形成、细胞分化等重要过程。线粒体也是最容易受损的一个细胞器,其可以显示细胞受损的程度。开发实时监测线粒体结构和功能指标变化的分析方法对于预防和诊断药源性线粒体损伤相关疾病具有重要的意义。
6.1 荧光成像技术 线粒体内含有各种生物活性物质,如活性氧、活性硫、活性氮、各种生物活性离子等,它们在人体各种生理和病理过程中发挥着重要作用,其含量水平的异常都可直接影响线粒体功能并导致脏器功能降低,严重时甚至危及生命。因此,精确检测线粒体内活性成分含量变化对于从分子水平上研究药物毒性与脏器功能之间的关系具有非常重要的生物医学意义。荧光标记技术作为一种可视化、实时、动态的演示技术,荧光探针在活细胞中具有高灵敏度、高空间分辨率和实时成像等优点,在研究线粒体活动过程中被广泛地应用,通过荧光成像技术研究药物对线粒体功能影响已成为荧光探针领域的热点。目前常用特异靶标荧光探针结合共聚焦荧光显微镜和流式细胞仪进行药物线粒体毒性评价。线粒体成像荧光探针主要对线粒体成像和对线粒体内各种活性物质成像,通过荧光成像技术对线粒体结构和功能进行研究,如线粒体形态变化、线粒体内环境(pH值、黏度、电位等)变化、线粒体内源性活性氧物质的检测、ATP、谷胱甘肽等。近年来,人们对各种用于检测线粒体功能的荧光探针进行了广泛的研究,靶向定位于线粒体,常见的线粒体功能评价探针有线粒体膜电位探针(JC10、罗丹明123,四甲基罗丹明甲酯)、Ca2+离子探针(Fluo-4)、ROS探针(DCFH-DA)、线粒体质量探针(MitoTracker Green FM,MitoTracker Deep Red FM)、GSH探针(mBCl)[86]。
传统的荧光成像技术虽然可以实现活细胞层面监测药物对线粒体功能的动态影响,但是存在检测通量低,操作繁琐等局限性。高内涵分析(HCA)是一种基于细胞表型分析的高效新药筛选技术,能够在保持细胞结构和功能完整的前提下,同时检测待测样品对细胞的形态、生长、周期、迁移、调亡、代谢途径及信号传导等方面的影响,从单一实验中获取大量有关信息,从而确定化合物的生物活性以及潜在毒性[87]。目前HCA能够实时监测体外肝细胞与毒性机制相关的多个重耍标志分子,包括细胞核的形态和数量、线粒体膜电位以及氧化应激状态、调亡与早期DNA损伤的变化等,是体外评价化合物潜在肝毒性、从机制上预测候选药物安全性的高效手段[88]。该方法能够弥补动物试验灵敏度低、试验周期长,不能量化的缺点,因此,高通量毒性评价技术己成为药品质量风险与安全风险评估的新的技术趋势。多指标同步量化对药物的毒性进行了较为全面的分析,并可对其毒性作用机制做出初步的判断。
6.2 生化技术 线粒体是细胞的能量源,利用氧化磷酸化(OXPHOS)产生细胞约95%的ATP[9]。这一过程是由氧化磷酸化复合物(I,II,IV和V)完成的。因此,OXPHOS复合物的抑制会导致线粒体毒性。目前,已经开发出基于细胞/组织的免疫捕获或传统的生化方法用于测定参与氧化磷酸化的蛋白复合物的活性[89]。线粒体结构复杂,包含13个多肽且都是参与氧化磷酸化的蛋白质亚基,由mtDNA编码并在线粒体核糖体上合成。位于线粒体内的所有其他蛋白质都是核DNA编码,在胞质核糖体上合成,然后输入到线粒体中。干扰mtDNA或mtDNA编码蛋白合成的药物会损害氧化磷酸化,导致细胞ATP供应减少。传统上,采用免疫蛋白印迹法和测流试纸免疫测定法进行这些化合物的检测,然而,这2种方法都不适合高通量筛选[90-91]。
线粒体具有复杂的多腔结构,是一种复杂的细胞器,存在于每一个普通细胞中。除了产生能量外,线粒体还是活性氧(ROS)的来源,并在许多过程的调控中发挥核心作用,如细胞凋亡或二级信使的存储[92]。生物传感器提供了一种创新的工具,用于测量生物体内代谢产物的动态变化。此外,生物传感器更具有底物特异性,并且由基因编码,且不会引起不同细胞对探针摄取的异质性问题[93])。而且,通过选择合适的启动子,可以在时间上或空间上限制传感器的表达。同样,空间表达可以在细胞内进行调节,通过将引导传感器的前导肽连接到特定的细胞器或隔室,或者通过产生与定位蛋白质融合的传感器嵌合体。因此,有可能测量单细胞的单个细胞器中的代谢物,而不是群体。目前,多种生物传感器已被广泛应用到线粒体中,用于检测线粒体功能。
6.3 细胞能量代谢 线粒体最重要的功能是通过OXPHOS为细胞产生能量,因此,线粒体健康的主要标志是呼吸作用[94]。传统上,利用克拉克电极或将线粒体和整个细胞的耗氧量进行体外检测,但是缺乏高通量,利用pH和O2敏探针检测胞外酸化率(糖酵解)及线粒体电子传递链的功能[95-96]。目前较新的方法(例如使用Seahorse细胞外通量分析仪的方法)可以测量多孔板(24和96孔)中的呼吸(耗氧量)[97]。这种仪器可测量高达10 dpf的完整活斑马鱼和幼虫的耗氧量与细胞外酸化,以及从器官中仔细解剖的器官幼体和成年斑马鱼。
6.4 代谢组学分析技术 代谢组学(Metabonomics/Metabolomics)是效仿基因组学和蛋白质组學的研究思想,对生物体内所有代谢物进行定量分析,小如细胞,复杂如动物或人,并寻找代谢物与生理病理变化的相对关系的研究方式,是系统生物学的组成部分[98-99]。目前,代谢组学在诊断线粒体毒性方面的应用已在涉及抑制脂肪酸β氧化的案例中得到证实。基于代谢组学的分析可以很容易地检测线粒体毒性或遗传病,而无需分离线粒体。近年来,定量磁共振成像代谢组学已被应用于体外药物诱导线粒体毒性的研究。这种方法被用于研究C2C12肌管的整体变化,这些肌管被氧化磷酸化复合物I抑制剂(鱼藤酮)或氧化磷酸化复合物III抑制剂(抗霉素A)处理[100]。
6.5 电镜 先进的测试表征技术是认识微观世界的重要手段,对于尺度和维度观的培养具有重要意义。不同于传统光学显微镜和电子显微镜,原子力显微镜是通过其探针原子与样品的表面原子的作用力来实现对样品表面形貌观测的。传统的光学显微镜和电子显微镜是通过放大微观结构,然后直观的看,但是分辨率有一定的局限性,导致无法观测表面的细微结构。而AFM是通过与样品表面的“触觉”响应来重现事物的形貌。
冷冻电镜技术是样品在低温条件下迅速冷冻后,通过低温电镜进行观察和处理数据。电子显微镜在线粒体中的应用主要包括:1)观察由线粒体调控的细胞凋亡,即观察凋亡基因蛋白复合体动态变化的结构解析过程;2)观察有机物在线粒体内膜上的呼吸作用,同时有多种呼吸链蛋白复合体共同参与,冷冻电镜技术可以高效对这类大分子复合物的结构解析。总之,冷冻透射电子显微镜主要用于高效率地观察与线粒体活性相关的蛋白复合体的结构解析[101]。
7 结论与展望
总之,线粒体作为细胞内重要细胞器,药物诱导线粒体功能异常通常与机体脏器损伤密切相关。高效监控线粒体功能与结构动态变化对于药物安全性评价及相应机体损伤机制研究意义重大。中药材成分极其复杂,毒性机制涉及多因素,且作用涉及多靶点多途径,各种成分相互影响、相互作用,使得中药毒性研究具有极大的挑战性。未来需要在传统的离体线粒体及完整细胞评价模型基础上,开发更具仿生性人源性器官评价模型,同时选择更加灵敏的线粒体毒性靶标,应用高通量高内涵筛选技术,进行规模化的中西药线粒体毒性评价。此外,线粒体过去被认为是在细胞内相互孤立的细胞能量供应中心,而实际上,其在细胞中相互连接形成网状结构,并通过不断的融合与分裂处于一定的动态平衡之中。对于研究药物致使的器官损伤,在评估线粒体功能时也应该考虑其他细胞器的功能,将细胞内部细胞器作为一个整体网络进行监测,将有助于理解药物引起各种损伤机制及病变过程。
参考文献
[1]Hargreaves IP,Al Shahrani M,Wainwright L,Heales SJ.Drug-Induced Mitochondrial Toxicity[J].Drug Saf,2016,39(7):661-674.
[2]Kühlbrandt W.Structure and function of mitochondrial membrane protein complexes[J].BMC Biol,2015,13:89.
[3]Pereira CV,Moreira AC,Pereira SP,et al.Investigating drug-induced mitochondrial toxicity:a biosensor to increase drug safety?[J].Curr Drug Saf,2009,4(1):34-54.
[4]Nadanaciva S,Will Y.New insights in drug-induced mitochondrial toxicity[J].Curr Pharm Des,2011,17(20):2100-2112.
[5]Tsiper MV,Sturgis J,Avramova LV,et al.Differential mitochondrial toxicity screening and multi-parametric data analysis[J].PLoS One,2012,7(10):e45226.
[6]陳宇征,吕文良.中药导致药物性肝损伤的机制研究进展[J].中国中医基础医学杂志,2015,21(11):1476-1478.
[7]Gourlain K,Amellal B,Ait Arkoub Z,et al.Quantitative analysis of human mitochondrial DNA using a real-time PCR assay[J].HIV Med,2003,4(3):287-292.
[8]Swiss R,Will Y.Assessment of mitochondrial toxicity in HepG2 cells cultured in high-glucose-or galactose-containing media[J].Curr Protoc Toxicol,2011,2(20):tx0220s49.
[9]Swiss R,Niles A,Cali JJ,et al.Validation of a HTS-amenable assay to detect drug-induced mitochondrial toxicity in the absence and presence of cell death[J].Toxicol In Vitro,2013,27(6):1789-1797.
[10]Sakamuru S,Attene-Ramos MS,Xia M.Mitochondrial Membrane Potential Assay[J].Methods Mol Biol,2016,1473:17-22.
[11]Rodrigues RM,Macko P,Palosaari T,et al.Autofluorescence microscopy:a non-destructive tool to monitor mitochondrial toxicity[J].Toxicol Lett,2011,206(3):281-288.
[12]Vuda M,Kamath A.Drug induced mitochondrial dysfunction:Mechanisms and adverse clinical consequences[J].Mitochondrion,2016,31:63-74.
[13]杨婷婷,江振洲,张陆勇.经由线粒体损伤诱发的药源性肝损伤研究进展[J].药学进展,2014,38(11):809-818.
[14]Knobeloch LM,Blondin GA,Read HW,et al.Assessment of chemical toxicity using mammalian mitochondrial electron transport particles[J].Arch Environ Contam Toxicol,1990,19(6):828-835.
[15]Turrens JF.Mitochondrial formation of reactive oxygen species[J].J Physiol,2003,552(2):335-344.
[16]Yakes FM,Van Houten B.Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress[J].Proc Natl Acad Sci USA,1997,94(2):514-519.
[17]Serviddio G,Bellanti F,Giudetti AM,et al.Mitochondrial oxidative stress and respiratory chain dysfunction account for liver toxicity during amiodarone but not dronedarone administration[J].Free Radic Biol Med,2011,51(12):2234-2242.
[18]Deavall DG,Martin EA,Horner JM,et al.Drug-induced oxidative stress and toxicity[J].J Toxicol,2012,2012:645460.
[19]Westphal D,Dewson G,Czabotar PE,et al.Molecular biology of Bax and Bak activation and action[J].Biochim Biophys Acta,2011,1813(4):521-531.
[20]Garrido C,Galluzzi L,Brunet M,et al.Mechanisms of cytochrome c release from mitochondria[J].Cell Death Differ,2006,13(9):1423-1433.
[21]Berson A,Schmets L,Fisch C,et al.Inhibition by nilutamide of the mitochondrial respiratory chain and ATP formation.Possible contribution to the adverse effects of this antiandrogen[J].J Pharmacol Exp Ther,1994,270(1):167-176.
[22]Moreno-Sánchez R,Bravo C,Vásquez C,et al.Inhibition and uncoupling of oxidative phosphorylation by nonsteroidal anti-inflammatory drugs:study in mitochondria,submitochondrial particles,cells,and whole heart[J].Biochem Pharmacol,1999,57(7):743-752.
[23]Miró O,López S,Pedrol E,et al.Mitochondrial DNA depletion and respiratory chain enzyme deficiencies are present in peripheral blood mononuclear cells of HIV-infected patients with HAART-related lipodystrophy[J].Antivir Ther,2003,8(4):333-338.
[24]Masubuchi Y,Yamada S,Horie T.Possible mechanism of hepatocyte injury induced by diphenylamine and its structurally related nonsteroidal anti-inflammatory drugs[J].J Pharmacol Exp Ther,2000,292(3):982-7.
[25]Masubuchi Y,Yamada S,Horie T.Diphenylamine as an important structure of nonsteroidal anti-inflammatory drugs to uncouple mitochondrial oxidative phosphorylation[J].Biochem Pharmacol,1999,58(5):861-5.
[26]O′Connor N,Dargan PI,Jones AL.Hepatocellular damage from non-steroidal anti-inflammatory drugs[J].QJM,2003,96(11):787-791.
[27]Berson A,Cazanave S,Descatoire V,et al.The anti-inflammatory drug,nimesulide(4-nitro-2-phenoxymethane-sulfoanilide),uncouples mitochondria and induces mitochondrial permeability transition in human hepatoma cells:protection by albumin[J].J Pharmacol Exp Ther,2006,318(1):444-454.
[28]Kalghatgi S,Spina CS,Costello JC,et al.Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells[J].Sci Transl Med,2013,5(192):192ra85.
[29]Pochini L,Galluccio M,Scumaci D,et al.Interaction of beta-lactam antibiotics with the mitochondrial carnitine/acylcarnitine transporter[J].Chem Biol Interact,2008,173(3):187-194.
[30]Soriano A,Miró O,Mensa J.Mitochondrial toxicity associated with linezolid[J].N Engl J Med,2005,353(21):2305-2306.
[31]Kakuda TN.Pharmacology of nucleoside and nucleotide reverse transcriptase inhibitor-induced mitochondrial toxicity[J].Clin Ther,2000,22(6):685-708.
[32]Simnek T,Stérba M,Popelová O,et al.Anthracycline-induced cardiotoxicity:overview of studies examining the roles of oxidative stress and free cellular iron[J].Pharmacol Rep,2009,61(1):154-171.
[33]Peres LA,da Cunha AD Jr.Acute nephrotoxicity of cisplatin:molecular mechanisms[J].J Bras Nefrol,2013,35(4):332-340.
[34]Miró O,Barrientos A,Alonso JR,et al.Effects of general anaesthetic procedures on mitochondrial function of human skeletal muscle[J].Eur J Clin Pharmacol,1999,55(1):35-41.
[35]Vanlander AV,Okun JG,de Jaeger A,et al.Possible pathogenic mechanism of propofol infusion syndrome involves coenzyme q[J].Anesthesiology,2015,122(2):343-352.
[36]Bosnjak ZJ,Yan Y,Canfield S,et al.Ketamine induces toxicity in human neurons differentiated from embryonic stem cells via mitochondrial apoptosis pathway[J].Curr Drug Saf,2012,7(2):106-119.
[37]Liu F,Patterson TA,Sadovova N,et al.Ketamine-induced neuronal damage and altered N-methyl-D-aspartate receptor function in rat primary forebrain culture[J].Toxicol Sci,2013,131(2):548-557.
[38]Sun X,Garlid KD.On the mechanism by which bupivacaine conducts protons across the membranes of mitochondria and liposomes[J].J Biol Chem,1992,267(27):19147-19154.
[39]Sztark F,Malgat M,Dabadie P,et al.Comparison of the effects of bupivacaine and ropivacaine on heart cell mitochondrial bioenergetics[J].Anesthesiology,1998,88(5):1340-1349.
[40]Irwin W,Fontaine E,Agnolucci L,et al.Bupivacaine myotoxicity is mediated by mitochondria[J].J Biol Chem,2002,277(14):12221-12227.
[41]Tolosa L,Carmona A,Castell JV,et al.High-content screening of drug-induced mitochondrial impairment in hepatic cells:effects of statins[J].Arch Toxicol,2015,89(10):1847-1860.
[42]Stringer HA,Sohi GK,Maguire JA,et al.Decreased skeletal muscle mitochondrial DNA in patients with statin-induced myopathy[J].J Neurol Sci,2013,325(1-2):142-147.
[43]Brunmair B,Lest A,Staniek K,et al.Fenofibrate impairs rat mitochondrial function by inhibition of respiratory complex I[J].J Pharmacol Exp Ther,2004,311(1):109-114.
[44]Piel S,Ehinger JK,Elmér E,et al.Metformin induces lactate production in peripheral blood mononuclear cells and platelets through specific mitochondrial complex I inhibition[J].Acta Physiol(Oxf),2015,213(1):171-180.
[45]Hu D,Wu CQ,Li ZJ,et al.Characterizing the mechanism of thiazolidinedione-induced hepatotoxicity:An in vitro model in mitochondria[J].Toxicol Appl Pharmacol,2015,284(2):134-141.
[46]Lheureux PE,Penaloza A,Zahir S,et al.Science review:carnitine in the treatment of valproic acid-induced toxicity-what is the evidence?[J].Crit Care,2005,9(5):431-440.
[47]Komulainen T,Lodge T,Hinttala R,et al.Sodium valproate induces mitochondrial respiration dysfunction in HepG2 in vitro cell model[J].Toxicology,2015,331:47-56.
[48]Santos NA,Medina WS,Martins NM,et al.Aromatic antiepileptic drugs and mitochondrial toxicity:effects on mitochondria isolated from rat liver[J].Toxicol In Vitro,2008,22(5):1143-1152.
[49]Cruz TS,Faria PA,Santana DP,et al.On the mechanisms of phenothiazine-induced mitochondrial permeability transition:Thiol oxidation,strict Ca2+ dependence,and cyt c release[J].Biochem Pharmacol,2010,80(8):1284-1295.
[50]Contreras-Shannon V,Heart DL,Paredes RM,et al.Clozapine-induced mitochondria alterations and inflammation in brain and insulin-responsive cells[J].PLoS One,2013,8(3):e59012.
[51]Lee MY,Hong S,Kim N,et al.Tricyclic Antidepressants Amitriptyline and Desipramine Induced Neurotoxicity Associated with Parkinson′s Disease[J].Mol Cells,2015,38(8):734-740.
[52]Hroudová J,Fiar Z.In vitro inhibition of mitochondrial respiratory rate by antidepressants[J].Toxicol Lett,2012,213(3):345-352.
[53]Li Y,Couch L,Higuchi M,et al.Mitochondrial dysfunction induced by sertraline,an antidepressant agent[J].Toxicol Sci,2012,127(2):582-591.
[54]Dykens JA,Jamieson JD,Marroquin LD,et al.In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone,trazodone,and buspirone[J].Toxicol Sci,2008,103(2):335-345.
[55]Heidari R.The footprints of mitochondrial impairment and cellular energy crisis in the pathogenesis of xenobiotics-induced nephrotoxicity,serum electrolytes imbalance,and Fanconi′s syndrome:A comprehensive review[J].Toxicology,2019,423:1-31.
[56]Amacher DE.Drug-associated mitochondrial toxicity and its detection[J].Curr Med Chem,2005,12(16):1829-1839.
[57]Yao J,Jiang Z,Duan W,et al.Involvement of mitochondrial pathway in triptolide-induced cytotoxicity in human normal liver L-02 cells[J].Biol Pharm Bull,2008,31(4):592-597.
[58]Wang P,Pradhan K,Zhong XB,et al.Isoniazid metabolism and hepatotoxicity[J].Acta Pharm Sin B,2016,6(5):384-392.
[59]Walker UA,Bickel M,Lütke Volksbeck SI,et al.Evidence of nucleoside analogue reverse transcriptase inhibitor--associated genetic and structural defects of mitochondria in adipose tissue of HIV-infected patients[J].J Acquir Immune Defic Syndr,2002,29(2):117-121.
[60]Meyer JN,Leuthner TC,Luz AL.Mitochondrial fusion,fission,and mitochondrial toxicity[J].Toxicology,2017,391:42-53.
[61]Andrienko T,Kuznetsov AV,Kaambre T,et al.Metabolic consequences of functional complexes of mitochondria,myofibrils and sarcoplasmic reticulum in muscle cells[J].J Exp Biol,2003,206(Pt 12):2059-2072.
[62]Dykens JA,Will Y.The significance of mitochondrial toxicity testing in drug development[J].Drug Discov Today,2007,12(17-18):777-785.
[63]Hynes J,Will Y.The Evolution of Mitochondrial Toxicity Assessment in Industry[J].Mitochondrial Biology and Experimental Therapeutics,2018,3:319-332.
[64]Schulz S,Lichtmannegger J,Schmitt S,et al.A protocol for the parallel isolation of intact mitochondria from rat liver,kidney,heart,and brain[J].Methods Mol Biol,2015,1295:75-86.
[65]Wieckowski MR,Giorgi C,Lebiedzinska M,et al.Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells[J].Nat Protoc,2009,4(11):1582-1590.
[66]Frezza C,Cipolat S,Scorrano L.Organelle isolation:functional mitochondria from mouse liver,muscle and cultured fibroblasts[J].Nat Protoc,2007,2(2):287-295.
[67]Picard M,Taivassalo T,Gouspillou G,et al.Mitochondria:isolation,structure and function[J].J Physiol,2011,589(18):4413-4421.
[68]Yamada KM,Cukierman E.Modeling tissue morphogenesis and cancer in 3D[J].Cell,2007,130(4):601-610.
[69]An R,Merrill D,Avramova L,et al.Phenotypic profiling of Raf inhibitors and mitochondrial toxicity in 3D tissue using biodynamic imaging[J].J Biomol Screen,2014,19(4):526-537.
[70]Rossignol R,Gilkerson R,Aggeler R,et al.Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells[J].Cancer Res,2004,64(3):985-993.
[71]PIERCE WA Jr.Glucose and galactose metabolism in Streptococcus pyogenes[J].J Bacteriol,1957,74(2):186-193.
[72]Marroquin LD,Hynes J,Dykens JA,et al.Circumventing the Crabtree effect:replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants[J].Toxicol Sci,2007,97(2):539-547.
[73]Elkalaf M,Anděl M,Trnka J.Low glucose but not galactose enhances oxidative mitochondrial metabolism in C2C12 myoblasts and myotubes[J].PLoS One,2013,8(8):e70772.
[74]Rana P,Nadanaciva S,Will Y.Mitochondrial membrane potential measurement of H9c2 cells grown in high-glucose and galactose-containing media does not provide additional predictivity towards mitochondrial assessment[J].Toxicol In Vitro,2011,25(2):580-587.
[75]Xu Q,Liu L,Vu H,et al.Can Galactose Be Converted to Glucose in HepG2 Cells? Improving the in Vitro Mitochondrial Toxicity Assay for the Assessment of Drug Induced Liver Injury[J].Chem Res Toxicol,2019,32(8):1528-1544.
[76]Hamilton BF,Stokes AH,Lyon J,et al.In vivo assessment of mitochondrial toxicity[J].Drug Discov Today,2008,13(17-18):785-790.
[77]Mandal A,Pinter K,Drerup CM.Analyzing Neuronal Mitochondria in vivo Using Fluorescent Reporters in Zebrafish[J].Front Cell Dev Biol,2018,6:144.
[78]Robinson BL,Dumas M,Ali SF,et al.Mechanistic studies on ketamine-induced mitochondrial toxicity in zebrafish embryos[J].Neurotoxicol Teratol,2018,69:63-72.
[79]de Boer R,Smith RL,De Vos WH,et al.Caenorhabditis elegans as a Model System for Studying Drug Induced Mitochondrial Toxicity[J].PLoS One,2015,10(5):e0126220.
[80]Raftery TD,Jayasundara N,Di Giulio RT.A bioenergetics assay for studying the effects of environmental stressors on mitochondrial function in vivo in zebrafish larvae[J].Comp Biochem Physiol C Toxicol Pharmacol,2017,192:23-32.
[81]Aryaman J,Johnston IG,Jones NS.Mitochondrial Heterogeneity[J].Frontiers in Genetics,2019,9:22-45.
[82]Boehning AL,Essien SA,Underwood EL,et al.Cell type-dependent effects of ellagic acid on cellular metabolism[J].Biomed Pharmacother,2018,106:411-418.
[83]Ngai KC,Yeung CY,Leung CS.Difference in susceptibilities of different cell lines to bilirubin damage[J].J Paediatr Child Health,2000,36(1):51-55.
[84]Aryaman J,Hoitzing H,Burgstaller JP,et al.Mitochondrial heterogeneity,metabolic scaling and cell death[J].Bioessays,2017,39(7):28594445.
[85]Maclennan RA,Eakins J,Bauch C,et al.A comprehensive comparison of in vitro assays utilised to detect mitochondrial toxicity[J].Toxicology Letters,2018,295:S200.
[86]Hoornstra D,Andersson MA,Mikkola R,et al.A new method for in vitro detection of microbially produced mitochondrial toxins[J].Toxicol In Vitro,2003,17(5-6):745-751.
[87]Attene-Ramos MS,Huang R,Sakamuru S,et al.Systematic study of mitochondrial toxicity of environmental chemicals using quantitative high throughput screening[J].Chem Res Toxicol,2013,26(9):1323-1332.
[88]Wills LP.The use of high-throughput screening techniques to evaluate mitochondrial toxicity[J].Toxicology,2017,391:34-41.
[89]Nadanaciva S,Dykens JA,Bernal A,et al.Mitochondrial impairment by PPAR agonists and statins identified via immunocaptured OXPHOS complex activities and respiration[J].Toxicol Appl Pharmacol,2007,223(3):277-287.
[90]Nagiec EE,Wu L,Swaney SM,et al.Oxazolidinones inhibit cellular proliferation via inhibition of mitochondrial protein synthesis[J].Antimicrob Agents Chemother,2005,49(9):3896-3902.
[91]Nadanaciva S,Willis JH,Barker ML,et al.Lateral-flow immunoassay for detecting drug-induced inhibition of mitochondrial DNA replication and mtDNA-encoded protein synthesis[J].J Immunol Methods,2009,343(1):1-12.
[92]Brookes PS,Yoon Y,Robotham JL,et al.Calcium,ATP,and ROS:a mitochondrial love-hate triangle[J].Am J Physiol Cell Physiol,2004,287(4):C817-833.
[93]Jones AM,Grossmann G,Danielson J,et al.In vivo biochemistry:applications for small molecule biosensors in plant biology[J].Curr Opin Plant Biol,2013,16(3):389-395.
[94]Prill S,Bavli D,Levy G,et al.Real-time monitoring of oxygen uptake in hepatic bioreactor shows CYP450-independent mitochondrial toxicity of acetaminophen and amiodarone[J].Arch Toxicol,2016,90(5):1181-1191.
[95]Hynes J,Natoli E Jr,Will Y.Fluorescent pH and oxygen probes of the assessment of mitochondrial toxicity in isolated mitochondria and whole cells[J].Curr Protoc Toxicol,2009,2(16):tx0216s40.
[96]Hynes J,Nadanaciva S,Swiss R,et al.A high-throughput dual parameter assay for assessing drug-induced mitochondrial dysfunction provides additional predictivity over two established mitochondrial toxicity assays[J].Toxicol In Vitro,2013,27(2):560-569.
[97]Beeson CC,Beeson GC,Schnellmann RG.A high-throughput respirometric assay for mitochondrial biogenesis and toxicity[J].Anal Biochem,2010,404(1):75-81.
[98]Nicholson JK,Connelly J,Lindon JC,et al.Metabonomics:a platform for studying drug toxicity and gene function[J].Nat Rev Drug Discov,2002,1(2):153-161.
[99]Xu EY,Schaefer WH,Xu Q.Metabolomics in pharmaceutical research and development:metabolites,mechanisms and pathways[J].Curr Opin Drug Discov Devel,2009,12(1):40-52.
[100]Xu Q,Vu H,Liu L,et al.Metabolic profiles show specific mitochondrial toxicities in vitro in myotube cells[J].J Biomol NMR,2011,49(3-4):207-219.
[101]杨宁宇.冷冻电镜技术在线粒體中的应用研究[J].中国高新科技,2019,3(3):100-102.
(2020-11-19收稿 责任编辑:王明)
