基于多头注意力机制和残差神经网络的肽谱匹配打分算法
2020-06-20王海鹏牟长宁
闵 鑫,王海鹏,牟长宁
(山东理工大学计算机科学与技术学院,山东淄博 255000)
(∗通信作者电子邮箱hpwang@sdut.edu.cn)
0 引言
现如今主流的蛋白质序列鉴定方法中,序列数据库搜索法[1]因其对不同的质谱类型表现出很强的鲁棒性且性能良好而成为最常用的鉴定方法。序列数据库搜索法的目的是在蛋白质序列数据库中搜索匹配出最可能产生给定实验质谱的序列,具体步骤一般分为三部分:对于每一张实验质谱,首先,从序列数据库中提取出与产生该实验质谱的母离子质量偏差在一定范围内的所有肽段形成肽谱匹配;然后,计算出每一个候选肽段的理论质谱和对应的实验质谱的得分;最后,再根据分数对肽谱匹配进行排序,取出分数最高者作为鉴定结果。其中,有效准确地计算出肽谱匹配得分对最终的鉴定结果起着决定性的作用。大多数打分算法都依赖于将候选肽转换成理论质谱,然后计算理论质谱与实验质谱的某种相似度分数,再通过全局信息进行二次打分。目前比较新的常用数据库搜索鉴定工具主要包括Comet[2-3]、MSGF+[4]、Mascot[5]、MaxQuant[6]等,其中Comet 和MSGF+作为代表性的开源鉴定工具被广泛使用,但是这些传统的鉴定工具中所使用的肽谱匹配打分方法都依赖于已知的肽碎裂规律及相关信息,存在着理论质谱准确度和相似度计算方法有效性等限制。
使用非深度学方法解决肽谱匹配打分问题一直是一个备受关注的问题,Bai 等[7]使用最大二分匹配模型进行肽谱匹配打分并取得了不错的效果,但由于最大二分匹配模型存在的问题导致肽谱匹配打分仍旧存在很大的问题。为改进此类问题,Bai 等[8]又提出了子模块广义匹配(Submodular Generalized Matching,SGM)模型,该模型在匹配准确率上有一定的提升,但是其充分利用质谱离子峰所携带的信息以及肽碎裂规律的能力还略显不足。深度学习领域不断的发展与完善,也为解决蛋白组学中的一些问题提供了新的途径。许多深度学习模型被用来解决蛋白组学的一些重要问题,如利用深度学习预测蛋白质结构[9-10]。蛋白组学中的许多问题可以看作是序列信息处理的延伸和拓展,而序列信息处理在深度学习领域中也是一个备受关注的问题。循环神经网络常常被用作序列信息处理,Zhou 等[11]提出了基于双向长短期记忆网络(Bidirectional Long Short-Term Memory network,Bi-LSTM)的碎片离子预测模型用于预测理论质谱,该模型具有良好的预测性能,但是并未进行进一步的肽谱匹配打分实验。随着深度学习的发展,众多学者发现ResNet(Residual neural Network)[12]不仅能有效解决计算机视觉领域许多重要问题,也能被用来有效地处理序列信息,并且已经有一些学者成功将ResNet应用到了蛋白组学的相关领域[13-14]。在现今的自然语言处理领域,许多取得不错效果的深度学习模型都使用注意力机制[15],注意力机制的核心目标是从众多信息中选择出对当前任务目标更关键的信息加以利用。在肽碎裂事件中,某一肽键的断裂不只与相邻两个氨基酸相关,也会与其他位置的氨基酸存在一定的关联,因此利用注意力机制能有效地利用这样一些相关性,进而提高模型的准确度。而多头自注意力机制(multi-head self-attention mechanism)[16]是对普通注意力机制的改进。因此本文提出了基于深度学习的打分算法:deepScore-α。该算法使用ResNet 与多头注意力机制相结合的模型用于肽谱匹配打分,有效地结合了两者优点,利用深度学习自动学习肽碎裂相关规律,能获得比较好的打分效果。
1 模型与算法
1.1 deepScore-α算法打分流程及模型结构
deepScore-α 打分算法使用深度学习模型学习肽碎裂规律对肽谱匹配进行打分,输入为肽谱匹配(Peptide Spectrum Match,PSM)中肽序列以及该肽序列在对应谱图中已标注的碎片离子离散化相对强度。算法分为两个阶段,输入的肽序列通过特征提取算法转换为特征序列与肽序列对应的碎片离子离散化相对强度一同输入模型,不同阶段输入相同,但是会产生不同的输出(具体流程如图1所示)。
1)在训练阶段,模型输出预测的碎裂离子离散化相对强度以评估模型学习肽碎裂规律的效果,并使用交叉熵作为模型损失进行权重更新。
2)在打分阶段,模型输出肽序列对应的碎片离子相对强度概率分布矩阵,再结合实际的碎片离子相对强度计算最终的肽谱匹配分数,而不是给出预测的碎片离子相对强度。
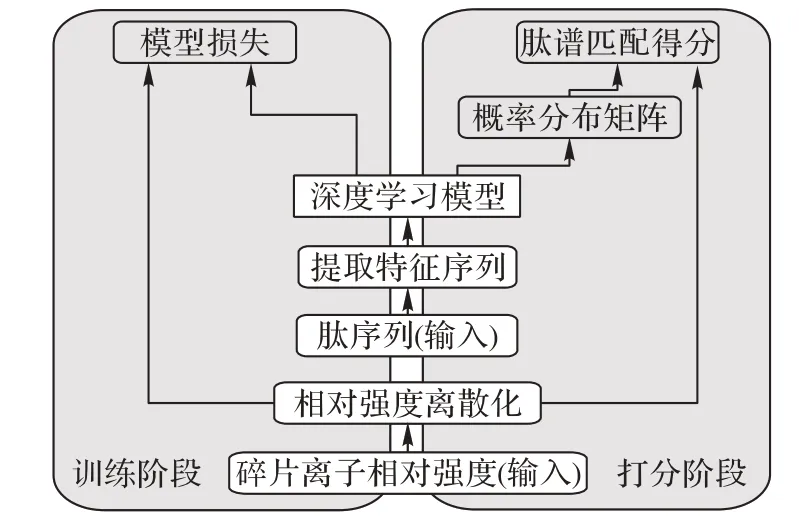
图1 deepScore-α打分算法流程Fig.1 Flowchart of deepScore-α scoring algorithm
deepScore-α 使用的模型由一维ResNet 模型和多头自注意力层组成:ResNet由卷积层和残差链接构成,主要用于提取和处理序列特征,多头自注意力机制则利用注意力机制的特性更加全面准备地学习肽碎裂规律以提升模型预测效果。ResNet 是深度卷积网络的改进,深度卷积网络随着网络层数的不断增加,在网络能够收敛的前提下,由于网络优化问题其表现会出现下降。ResNet通过残差连接能将较浅的网络层的输出直接输入到较深的网络层,在一定程度上解决了这个问题。deepScore-α 中所使用的ResNet20 参考了He 等[12]提出的ResNet,并且在此基础上进行了一些改进以适应肽序列信息处理,将其改为一维卷积且控制输入输出长度保持不变。
多头自注意力机制由谷歌于2017 年提出,不同于之前的注意力机制,多头自注意力机制采用了更加新颖的multi-head机制,即多次计算的注意力结果进行拼接,再通过线性变换获得最终的多头注意力结果。在长距离依赖上,因为自注意力需要计算每个输入位点与其他所有位点之间的关联,所以能够忽略位点之间的距离影响从而完整地学习到一个序列的内部结构信息。本文对多头注意力层中头的数量对最终模型预测结果的影响进行了比较实验,选取能产生最优实验结果的结构作为deepScore-α 最终的模型结构。本文所使用的multihead self-Attention ResNet20 如图2 所示,最终输出的b、y 代表b离子和y离子的离散化相对强度。
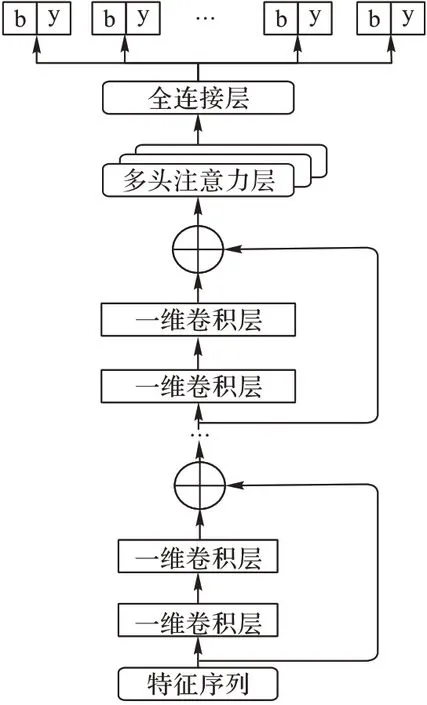
图2 deepScore-α模型结构Fig.2 Structure of deepScore-α model
1.2 deepScore-α分数计算方法
deepScore-α 所使用的打分算法是一种基于概率和的打分方法。假设候选肽氨基酸序列为X={A1,A2,…,An}(其中n为肽序列总长度),通过特征提取后获得对应的特征向量F={x1,x2,…,xn-1}。将特征向量序列作为输入,通过模型获得概率矩阵P。使用候选肽序列在谱图中进行谱峰标注,获得b离子及y 离子的相对谱峰强度N={n1,n2,…,no}(o为该肽序列产生的碎片离子总数),再通过以下公式进行离散化:

得到最终b/y 离子对应的离散化相对强度序列Y={y1,y2,…,yo}。使用以下公式计算出最终的肽谱匹配得分:
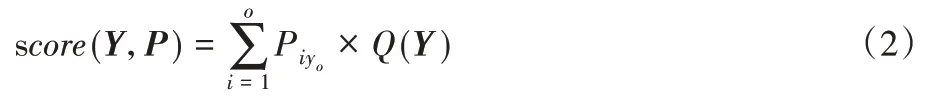
其中:n为肽序列产生的所有b/y离子总数;P为模型输出的碎片离子相对强度分布概率矩阵,Piyo表示第i个碎片离子实际的离散化相对强度yo在概率矩阵中对应的概率;Q(Y)为肽谱匹配质量系数计算函数。Q(Y)具体计算式如下:
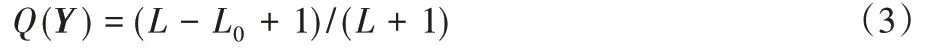
其中:L为离散化相对强度序列Y的长度,L0为其序列中相对强度为0的数量。
1.3 Comet及MSGF+打分算法
本文使用开源鉴定工具Comet以及MSGF+作为对比。其中,Comet 工具所采用的打分算法将实验质谱谱图转换为稀疏矩阵,并使用了快速交叉关联(Fast Cross Correlation)算法来计算实验质谱和理论质谱的相似度得分,再通过得到的相似度得分结合全局信息计算出e-value 作为最终肽谱匹配得分;MSGF+工具所采用的打分算法则将肽序列转化为肽序列向量,再使用概率模型将质谱转换为质谱向量,通过计算肽序列向量与质谱向量的点乘给出最终的肽谱匹配得分。以上两种鉴定工具所使用的打分算法均可看作是一种计算肽序列与实验质谱相似度的打分算法,本文提出的deepScore-α 则通过深度学习模型获得肽序列对应的碎片离子相对强度的概率和进行打分,无需计算肽序列与实验质谱的相似度,打破了相似度计算方法的限制。
2 实验与结果分析
2.1 数据集介绍及处理
本文用于模型训练的数据集为人类蛋白组数据集(HumanProteome),来自Wilhelm 等[17]在2014年关于人体蛋白组鉴定的相关工作(PRIDE 数据集标识符:PXD000865),包含人体26 个组织的蛋白质二级串联质谱谱图及相应的鉴定结果。在一般的肽序列鉴定流程中,蛋白质样品首先通过质谱仪获得原始质谱谱图数据(数据文件格式通常为raw),再使用相应的鉴定工具给出谱图的对应得分最高的肽段作为鉴定结果。由于鉴定软件给出的鉴定结果仍会存在错误,于是本文通过设置阈值条件q-value≤0.001 及PIF≥0.7 对鉴定结果进行过滤,并且在谱图存在多个鉴定结果时选取后验错误率(Posterior Error Probability,PEP)较低者作为最终的鉴定结果以保证鉴定结果的可信度(q-value、蛋白质水解诱导因子(Proteolysis Inducing Factor,PIF)和PEP 均为相应的统计学指标)。碰撞能量(Norm Collision Energy,NCE)为质谱仪器一种重要参数,本文使用碰撞能量对该数据集进行划分,为确保模型最终的训练效率,去除掉数据量过少的碰撞能量为40 的部分,最终获得两个训练数据集:790 271 条NCE 为30 的肽序列数据集,101 847 条NCE 为35 的肽序列数据集。随后使用标注误差限(碎片离子理论质荷比与实际谱图种谱峰质荷比的差值)为20 ppm 的条件对过滤处理后得到的数据集进行标注,获得肽序列对应的碎片离子谱峰强度,肽段碎裂会产生多种碎片离子,本文只考虑占比较大的b/y型碎片离子。
在进行打分效果评估时,对所有的原始谱图文件(raw)进行抽样,并利用pParse(v2015,一种谱图提取工具)提取出抽取得原始谱图文件中的谱图,再使用Comet(v2018014)和MSGF+(v2019.07.03)对提取的谱图进行鉴定。由于本文只针对肽谱匹配打分算法进行研究,所以为保证算法效果评估的有效性,在进行打分时对鉴定工具输出的每张谱图的前50个较高分数候选肽段进行重新打分。在进行打分Top1 命中率(在已知正确肽序列的情况下正确肽序列在该谱图所有候选肽中得分最高的谱图所占比例)实验时,为了保证命中率计算结果的准确性,分别从碰撞能量为30和35的模型测试集中抽取10 000张原始鉴定结果可信度比较高的谱图进行打分实验。用于算法泛化性能测试的数据集来自ProteomeTools2(PRIDE 数据集标识符:PXD010595)[18],同样对所有原始谱图数据进行随机抽样,再使用Comet和MSGF+进行鉴定,最后使用deepScore-α 对Comet 以及MSGF+输出的候选肽段进行重新打分并评估打分结果,由于ProteomeTools2数据集原始鉴定结果较为准确,所以直接进行Top1命中率评估。
2.2 特征提取及模型训练结果
在自然语言处理(Natural Language Processing,NLP)中,对单个字或词进行编码是十分重要的,因此逐渐发展出了WordEmbedding[19-20]这样的特征编码方式。而本文所涉及的肽碎裂事件中肽键可以被看作一个基本单位,即可以借鉴自然语言处理中的编码方式对其进行特征编码,通过类似onehot 类型的编码将氨基酸残基表示成22 维的向量。在肽键碎裂过程中,肽键左右相邻的氨基酸种类及肽序列中碱性氨基酸和肽序列所带电荷的数量对肽键的碎裂起着至关重要的作用。本文综合考虑了以上因素,最终形成了以下105 维的特征集(以某一肽键为例),具体特征集如表1所示。

表1 肽键特征集Tab.1 Feature set of peptide bonds
本文也探索了多头自注意力机制中头(head)的数量对最终模型的准确率产生的影响,在人类蛋白组数据集上分别进行了不同head 数量的实验,并且与逻辑回归以及支持向量机(Support Vector Machine,SVM)进行了比较,实验结果如表2所示。实验结果表明,本文采用的模型预测准确率明显优于逻辑回归和支持向量机,更有效地学习到了肽碎裂规律,且在不加入多头注意力机制时ACC(ACCuracy,预测的相对强度值与真实相对强度值相等的样本占所有样本的比例)为81.56%;加入多头注意力机制后,随着head数量的增加,模型预测结果逐步提升;当head 为8 时,模型准确率达到最优,相较于未加入注意力机制的测试结果提升了约2.6 个百分点;随着head 数量的进一步增多,模型准确率出现下降,于是deepScore-α后续采用了head为8的模型进行打分实验。

表2 不同模型在测试集上的预测准确率Tab.2 Prediction accuracy of different models on test set
模型训练中学习率设置为0.000 1,使用Adam 优化器,为避免模型过拟合,权重衰减设置为0.000 1,最终在测试集上测试结果如图3 及图4 所示。因为模型在训练时预测的是离散化的碎片离子相对强度,所以预测值与真实值相差大小为±1 时也可以认为预测有效。因此在评估模型预测效果时,除了将传统的ACC 作为评价标准,还需要考虑到预测值与真实值偏差范围为±1 的部分,从测试结果可以看出,模型在两个数据集上均获得了不错的预测效果,ACC 与±1 的部分所占比例之和都达到了97%。
2.3 肽谱匹配质量系数
对于某一实验质谱谱图,产生该谱图的正确肽序列相较于其他错误的候选肽序列往往能在谱图中标注出更多的非零相对强度的离子峰,有更多的相对强度不为零的碎片离子存在,即非零相对强度的占该肽序列产生的所有碎片离子数量的比例更大。由于deepScore-α 利用碎片离子离散化相对强度的概率和对候选肽进行打分,因此当正确肽序列长度短于错误候选肽序列时,即使错误候选肽序列有更多的相对强度为零的碎片离子,其概率和也可能超过正确肽序列。本文提出了肽谱匹配质量系数(PSM Quality Coefficient,PQC)用于降低肽序列长度及谱峰相对强度为零的碎片离子对打分算法的影响。
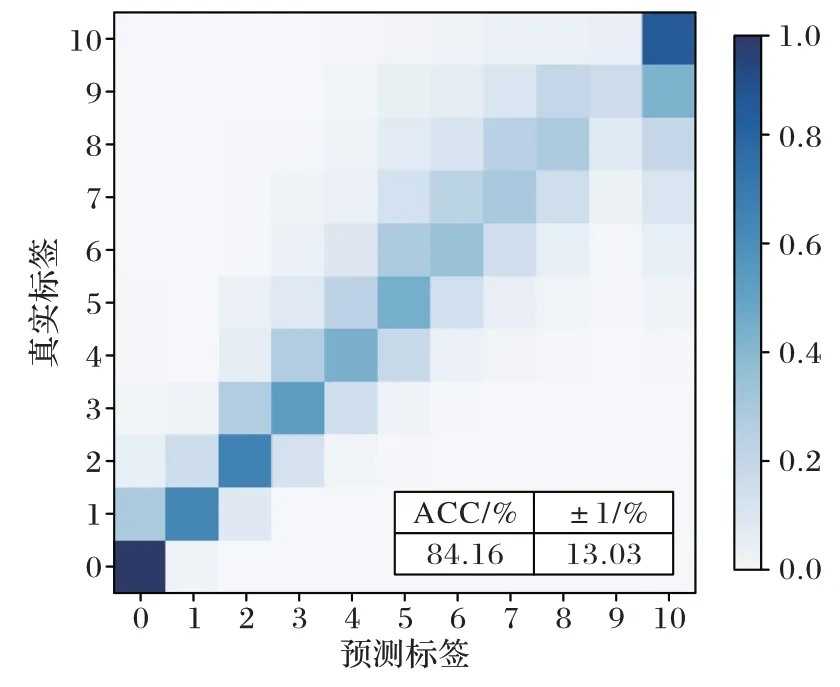
图3 碰撞能量为30的数据集上模型训练结果Fig.3 Model training results on dataset with NCE of 30
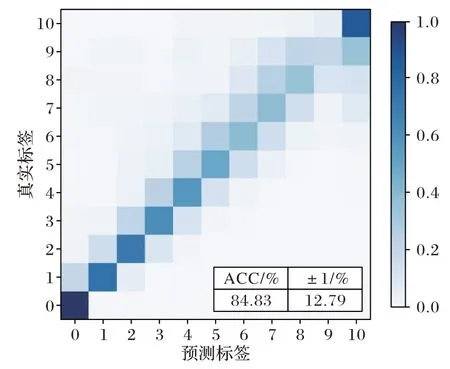
图4 碰撞能量为35的数据集上模型训练结果Fig.4 Model training results on dataset with NCE of 35
2.4 deepScore-α与Comet以及MSGF+的打分效果比较
本文使用Comet和MSGF+对碰撞能量为30及35的数据集中的随机抽取的原始数据文件(raw)进行鉴定,再使用deepScore-α对Comet以及MSGF+输出的前50条鉴定结果进行重新打分并排序,最终的错误发现率(False Discovery Rate,FDR)曲线如图5和图6所示。通过比较可以看出,deepScore-α在FDR=0.01时保留的肽序列数量相较于Comet和MSGF+最高提升了约14%,在30碰撞能量的情况下完全优于Comet和MSGF+,而在35碰撞能量的情况下某些部分基本与Comet和MSGF+持平。其原因应该是35碰撞能量的训练数据过少导致,30碰撞能量最终用于训练测试的包含790271条肽序列,而35碰撞能量训练集只有101847条肽序列,并且由于deepScore-α是利用概率进行打分,其对标签分类的概率更加敏感,因此交叉熵(Cross Entropy)更能反映模型对肽碎裂规律的学习程度。在最终用于打分的模型中,碰撞能量30的模型在测试集上的最小交叉熵为35.92,而在碰撞能量35中测试集的最小交叉熵为40.84,所以在利用概率进行打分时,deepScore-α在35碰撞能量的数据集上的表现比30碰撞能量差一些,如果能有效地扩大35碰撞能量的数据集规模,使其与30碰撞能量的数据集规模一致,使交叉熵降低至相同水平,其最终打分表现也应该一致。
在打分阶段比较打分算法有效分辨正确候选肽与错误候选肽的能力时,评估比较其Top1 命中率是十分有必要的。本文从划分为模型测试的数据集中随机抽取10 000 张谱,再使用Comet 和MSGF+对这10 000 张谱图进行重新打分,deepScore-α 对Comet 和MSGF+输出前50 的鉴定结果进行重新打分排序,最终的Top1 命中率比较如图7 所示。经过比较发现,deepScore-α 的Top1 命中率在30 碰撞能量以及35 碰撞能量的情况下都比Comet 和MSGF+更高,在30 碰撞能量的情况下更高出了5 个百分点,因此deepScore-α 识别正确肽序列的能力要强于Comet和MSGF+。

图5 deepScore-α、Comet和MSGF+在碰撞能量为30的人类蛋白组数据集上的FDR曲线Fig.5 FDR curves of deepScore-α,Comet and MSGF+on humanbody preteome dataset with NCE of 30

图6 deepScore-α、Comet和MSGF+在碰撞能量为35的人类蛋白组数据集上的FDR曲线Fig.6 FDR curves of deepScore-α,Comet and MSGF+on humanbody preteome dataset with NCE of 35
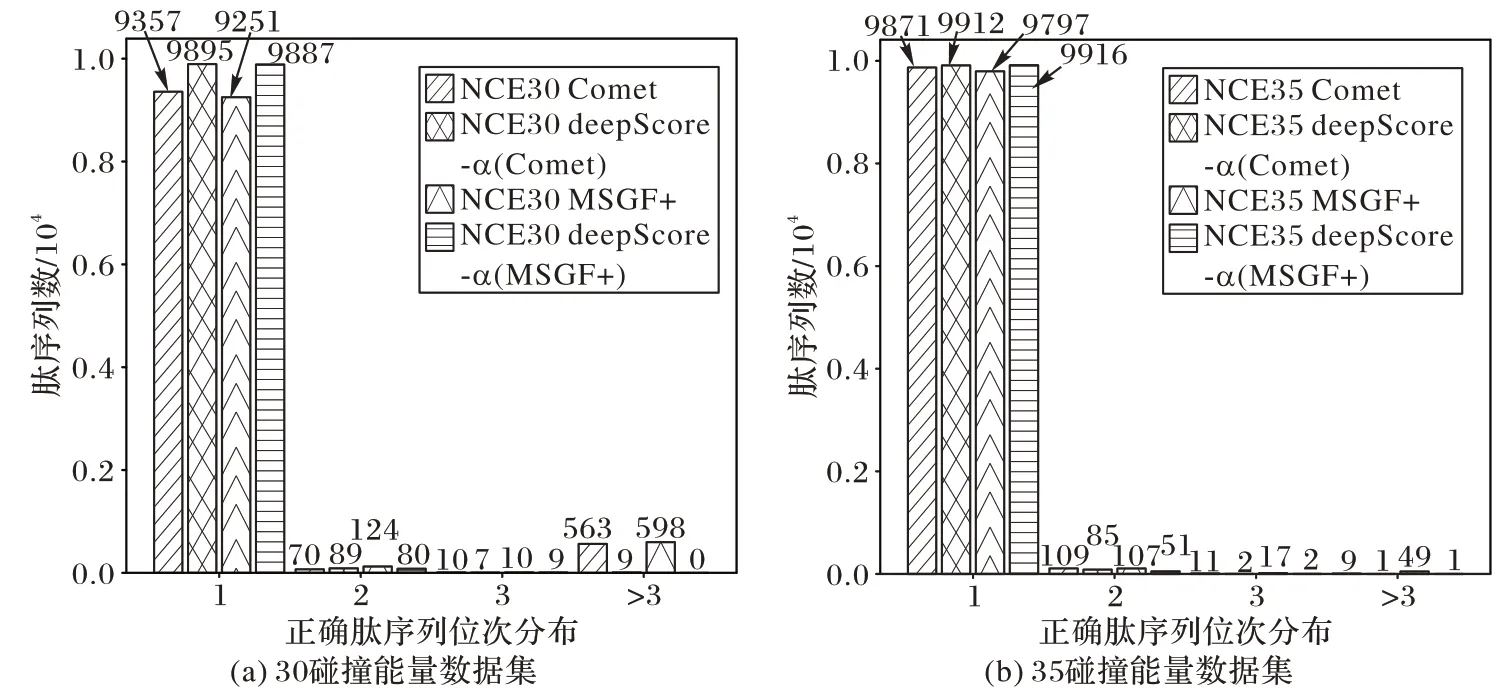
图7 deepScore-α、Comet和MSGF+打分结果中正确候选肽的分布Fig.7 Distribution of correct candidate peptides in deepScore-α,Comet and MSGF+scoring results
2.5 deepScore-α泛化性能分析
打分算法的泛化性能是十分重要的,好的泛化性能能极大地提高打分算法在不同数据集中的表现而不只是在特定某一数据集下表现良好。本文首先在Humanbody数据集上训练模型,再使用训练完成的模型对ProteomeTools2 中随机抽取的谱图进行打分并与Comet和MSGF+进行比较以评估其泛化性能。同样先使用Comet 和MSGF+进行鉴定,再使用deepScore-α 对二者输出的前50 条鉴定结果进行重新打分和排序,FDR 曲线如图8 和图9 所示。通过分析比较发现,deepScore-α 在FDR=0.01 条件下保留的肽序列相较于Comet和MSGF+最高提升了约7%,在30 碰撞能量下完全优于Comet,在35 碰撞能量下存在部分与Comet 基本持平的情况,与在Humanbody 数据集中进行打分的表现一致,应该为35 碰撞能量数据量相较30 碰撞能量不足所致。综合来看,deepScore-α 具有较好的泛化性能,利用Humabody 数据集训练的模型最终在ProteomeTools2 上的打分效果基本与原数据集上的一致,都优于Comet 和MSGF+。同样的,打分算法的Top1 的命中率也是其性能评价的一个重要指标。本文也评估了deepScore-α 在ProteomeTools2 数据上的Top1 命中率,实验结果如表3所示,deepScore-α 在ProteomeTools2数据集上的Top1 命中率相较于另外两个鉴定工具均提高了5 个百分点,Top1的鉴定结果中Decoy 的数量与另外两个鉴定工具相比减少了60%左右,因此可以判断deepScore-α 在打分性能上优于Comet和MSGF+,且具有较好的泛化性能。

图8 deepScore-α、Comet和MSGF+在碰撞能量为30的Proteome Tools2数据集上的FDR曲线Fig.8 FDR curves of deepScore-α,Comet and MSGF+on Proteome Tools2 dataset with NCE of 30

图9 deepScore-α、Comet和MSGF+在碰撞能量为35的Proteome Tools2数据集上的FDR曲线Fig.9 FDR curves of deepScore-α,Comet and MSGF+on Proteome Tools2 dataset with NCE of 35
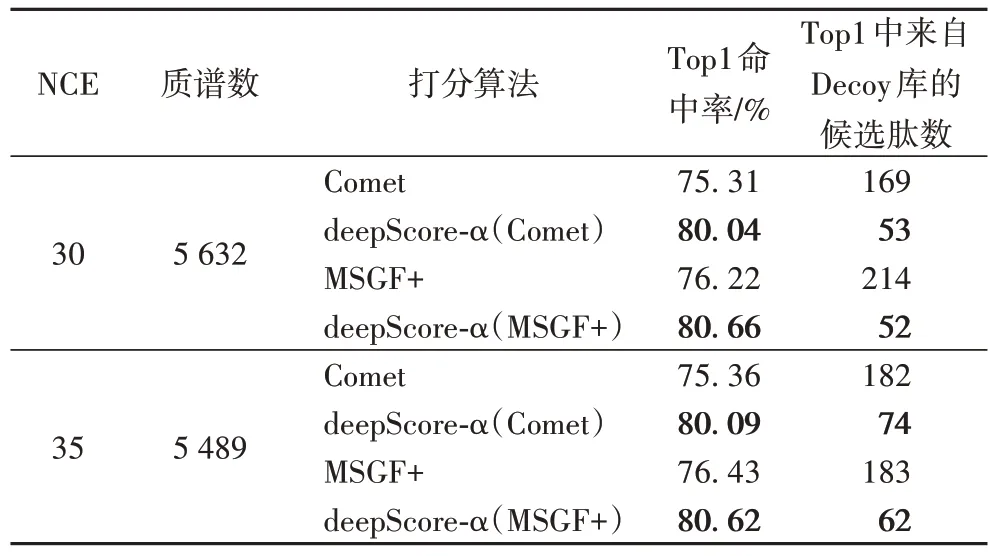
表3 deepScore-α、Comet和MSGF+在ProteomeTools2中打分结果比较Tab.3 Comparison of deepScore-α,Comet and MSGF+scoring results in ProteomeTools2
3 结语
deepScore-α 通过深度学习模型学习肽碎裂规律预测出碎片离子离散化相对强度分布概率,结合实际的碎片离子离散化相对强度形成肽谱匹配得分,有效地利用了深度学习的优势。deepScore-α 在人类蛋白组数据集上的表现优于常用开源鉴定工具Comet 和MSGF+,且使用人类蛋白组数据集训练的模型最终在ProteomeTools2 上的表现也优于另外两个鉴定工具,均可以证明deepScore-α 是一个打分效果优良且泛化能力较强的深度学习打分算法。下一步研究重点是拓展使用的碎片离子范围,本文在deepScore-α 打分算法中只使用了一价和二价的b/y离子,虽然在肽序列碎裂后产生的碎片离子中b/y离子占大多数,但是其产生诸如a/x、c/z离子以及相应的内部离子是否对打分算法产生影响还需要进一步验证。
