理论计算系列二苯乙烯化合物的电子结构与光谱性质*
2020-06-19杨明岸
杨明岸, 李 俊
(广东第二师范学院化学系,广东 广州 510800)
有机共轭化合物在疾病治疗、信号处理、太阳能电池和发光领域等的广泛应用, 引起了研究人员的重视,二苯乙烯化合物就是典型共轭结构的代表[1]。像紫檀芪、白藜芦醇等以二苯乙烯为母核的有机物在抗氧化与治疗肿瘤等方面的作用突出,表现出良好的生物活性,对肿瘤细胞生长的抑制效应显著[2]。二苯乙烯苷是中药何首乌的主要有效成分,可用于调节血脂和预防骨质疏松[3]。研究表明,二苯乙烯化合物一般具有较大的共轭长度和大的非线性极化率及优良的光电性能, 是一类很好的非线性光学生色团。近年来, 具有强双光子性能的有机材料在诸如双光子荧光显微术[4]、三维光存储[5]、三维微加工[6]、频率上转换激射[7]、光限幅[8]以及光动力疗法等[9]领域显示出良好的应用前景,双光子吸收材料结构与吸收的关系是当前国际研究的热点[10]。随着对强双光子吸收材料研究的深入,有机共轭发光材料技术的发展对发光材料的可调谐范围不断提出新要求。若要精细地改变前线轨道的能级,需要通过改变取代基的方式进行调节。然而目前关于取代基性质对发光分子电子结构及发光性能影响的研究并不多见,因此二苯乙烯化合物受到越来越多的关注[11]。合成这类化合物的关键在于两苯环之间乙烯桥的构建。通常以Wittig反应制备二苯乙烯类化合物,主要得到反式构型的产物[12]。事实上,人们还无法准确判断反应条件和取代基这两种因素对由Wittig反应得到的产物主要构型的影响。Hwang曾对不同取代的二苯乙烯化合物的Wittig合成结果进行过系统研究[13]。Pushkara和Breitung等通过理论计算,开展了杂环结构对二苯乙烯分子超极化率影响的相关研究[14]。从分子水平上看,有机分子的结构决定了其非线性光学特征。因此,深入研究分子结构上的差异对其非线性光学效应的影响, 归纳出分子结构与性质的规律,对未来材料科学的发展具有相当深远的意义。本文通过计算三种不同取代基型二苯乙烯类化合物的相关参数,考察其对电子结构和光谱性质的调控和影响,对化合物的实验设计与合成起到理论参考的作用。
1 理论计算
1.1 计算原理
1.2 计算方法
采用密度泛函理论(DFT),在B3LYP/3-21G*水平上,对化合物1~3(见图1~图3)进行了优化。以优化的几何结构为基础,进行了频率计算。计算结果表明,所有频率都为正值,表明优化得到的三个化合物的几何结构都是稳定结构。在获得的几何结构基础上,进行了分子轨道计算,获得了前线分子轨道能级。同时采用含时密度泛函理论(TDDFT)方法,在相同水平上计算了50个单重激发态,并对电子吸收光谱图进行了模拟,获得了三个化合物的电子吸收光谱图。所有计算均于量子化学软件包Gaussian 03完成。
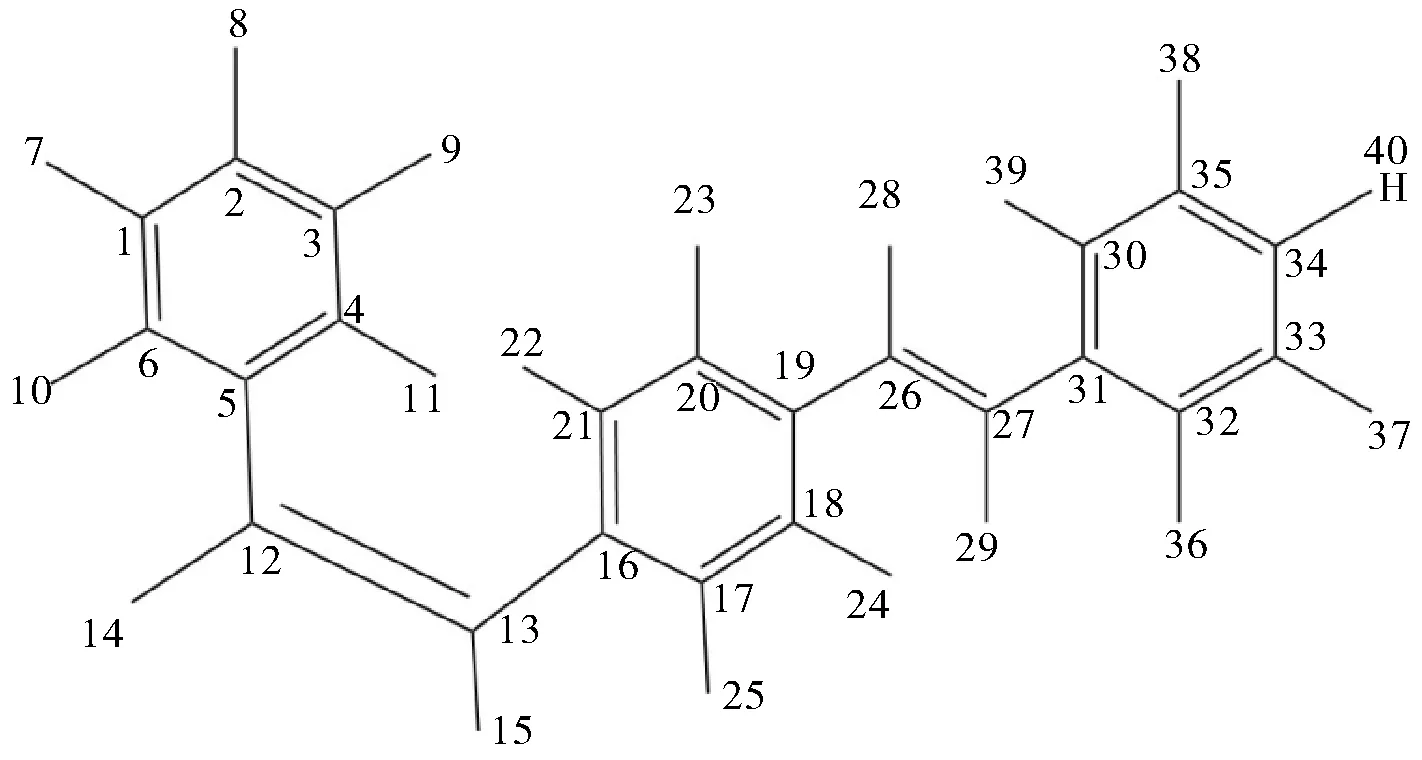
图1 化合物1的结构Fig.1 Structure of compound 1

图2 化合物2(硝基取代)的结构Fig.2 Structure of compound 2 (nitro substituted)

图3 化合物3(甲基取代)的结构Fig.3 Structure of compound 3 (methyl substituted)
2 结果与讨论
2.1 几何结构分析

表1 化合物1结构的主要几何参数Table 1 Main geometric parameters of the structure of compound 1

表2 化合物2(硝基取代)的主要几何参数Table 2 Main geometric parameters of compound 2(nitro substituted)

表3 化合物3(甲基取代)的主要几何参数Table 3 Main geometric parameters of compound 3 (methyl substituted)
二苯乙烯化合物的取代基不同,对物质结构的影响也不相同。我们选取了硝基和甲基取代的这三种不同取代基型二苯乙烯类化合物,分析其对键长、键角及二面角的影响。
采用密度泛函理论对三个化合物进行优化,获得了三个化合物的稳定几何结构,计算得到的几何参数如表1~表3所示。
由上述三个表格所列的数据可得,不同的取代基对二苯乙烯化合物的键长无影响,如C(26)-C(27)的键长,化合物1的为1.35 Å,化合物2的为1.35 Å,化合物3的为1.35 Å;以C(31)-C(27)-C(29)的键角为例,化合物1的为114.0°,化合物2的为116.0°,化合物3的为114.0°,其键角基本没有发生变化;以C(19)-C(26)-C(27)-C(31)的二面角为例,化合物1的为-7.1,化合物2的为-6.4,化合物3的为-7.6,其二面角仅存在小幅度变化。
2.2 前线分子轨道分析
前线分子轨道对探讨分子的结构与性质的关系具有重要作用,因此有必要对化合物前线分子轨道的组成和特征展开进一步的分析与讨论。对化合物1、化合物2和化合物3的最高占据轨道(HOMO)和次高占据轨道(SOMO)进行了记录,并计算出相应的能级差。三个化合物三重态的前线分子轨道组成和组合系数如表4所示。
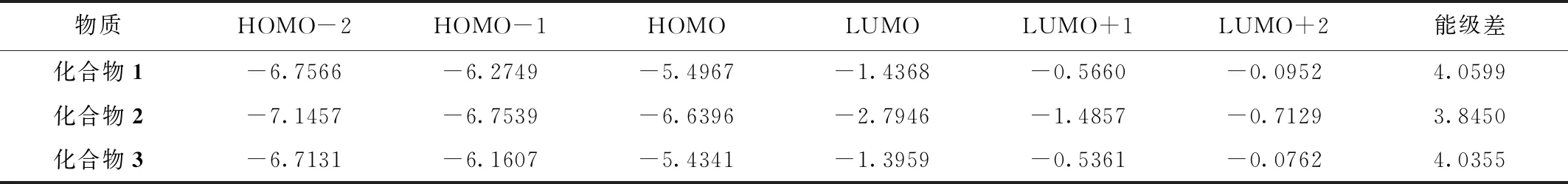
表4 三个化合物的前线分子轨道能级及能级差 Table 4 Frontier molecular orbital energy levels and energy level differences of the three compounds (eV)
从表4中可以发现,化合物1的能级差是4.0599 eV,化合物2的能级差是3.8450 eV,化合物3的能极差是4.0355 eV。由于能级差越小,波长越大。能级差大小排序为化合物1>化合物3>化合物2,故波长由长到短分别为化合物2、化合物3、化合物1。
2.3 电子吸收光谱分析
2.3.1 计算机程序Gaussian 03的模拟光谱图
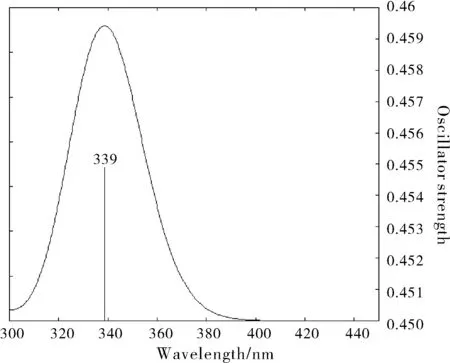
图4 化合物1的理论电子吸收光谱图Fig.4 Theoretical electron absorption spectrum of compound 1
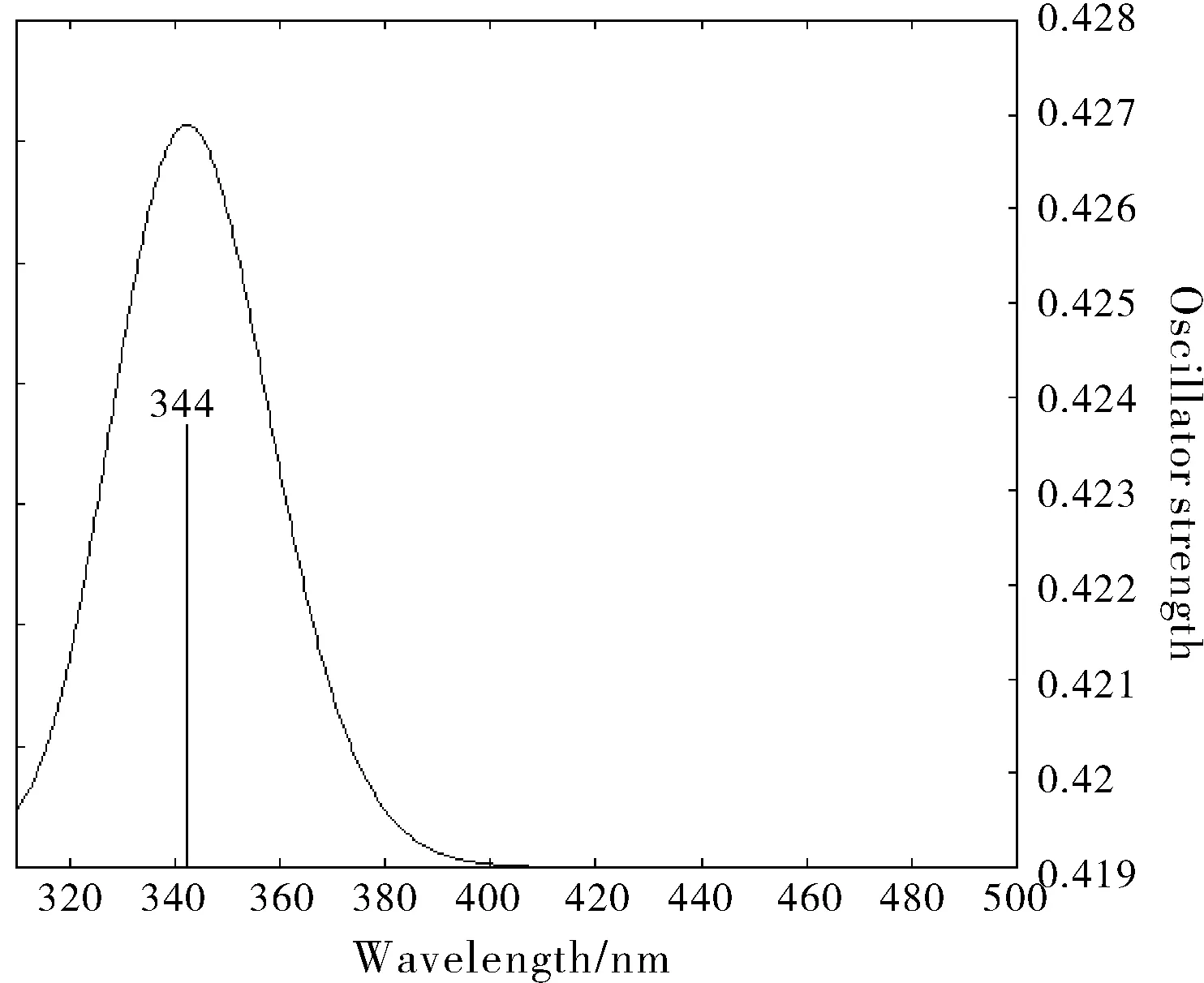
图5 化合物2(硝基取代)的理论电子吸收光谱图Fig.5 Theoretical electron absorption spectrum of compound 2 (nitro substituted)
2.3.2 实验测定的UV-vis吸收光谱图
图4~图6显示了三个化合物通过计算机模拟得到的在 300~500 nm 之间的电子吸收光谱,图7为三个化合物对应UV-vis测得的光谱图[13]。可见,在误差允许的范围内,波长由短至长的数据趋势一致,计算值与实验值基本一致。化合物1在吸收光谱中的最大吸收波长的理论值是339 nm,化合物2的是344 nm,化合物3的是342 nm,在该范围内产生的吸收是分子结构中共轭体系的π-π*跃迁所致。对于化合物3, 由于烷基的C-H 中σ电子与苯的π电子之间的σ-π超共轭效应, 有微弱的给电子能力,苯环的电子结构受到一定的影响, 故化合物3的电子吸收光谱较化合物1略有红移。化合物2属于生色团取代苯, 含有π键的生色团N=O与苯相连时产生π-π共轭, 形成更大的共轭体系, 因此电子吸收光谱较化合物1有较大程度的红移。

图6 化合物3(甲基取代)的理论电子吸收光谱图Fig.6 Theoretical electron absorption spectrum of compound 3 (methyl substituted)

图7 化合物1~3在实验中对应测得的UV-vis吸收光谱图Fig.7 Corresponding UV-vis absorption spectrum of compounds 1 to 3 measured in the experiment
3 结 论
本文用密度泛函理论与含时密度泛函理论的方法研究了化合物1、化合物2及化合物3这三种取代基型二苯乙烯类化合物,对它们在几何结构、电子结构和电子吸收光谱的性质三个方面分别进行了计算和分析。计算结果表明,不同的取代基对二苯乙烯类化合物的前线分子轨道能级和电子吸收光谱的影响较大, 电子吸收光谱均发生了红移现象。理论计算所得的数据与相应的实验数据基本吻合,波长变化的数据趋势相一致,具有较高的可信度。综上所述,本文在三种取代基型二苯乙烯类化合物的理论计算研究能够为分子的实验设计和合成提供有效的理论依据。
