MELK mRNA表达水平在非小细胞肺癌患者中的预后价值*
2020-06-16石旦徐斌蒋敬庭苏州大学附属第三医院肿瘤生物诊疗中心江苏省肿瘤免疫治疗工程技术研究中心苏州大学细胞治疗研究院江苏常州213003
石旦,徐斌,蒋敬庭(苏州大学附属第三医院肿瘤生物诊疗中心,江苏省肿瘤免疫治疗工程技术研究中心,苏州大学细胞治疗研究院,江苏常州213003)
目前已有多种针对非小细胞肺癌(NSCLC)特别是晚期NSCLC的分子靶向药物。但对于特定突变位点之外的NSCLC患者[1],仍迫切需要新的治疗靶点。本研究建立了一种基于预后价值的筛选生物标志物的方法,同时考虑候选基因在NSCLC良恶性组织中的表达差异,最终选取母体胚胎亮氨酸拉链激酶(MELK)作为研究对象。MELK又名小鼠蛋白丝氨酸/苏氨酸激酶38(MPK38)[2],是一种AMPK相关的丝氨酸—苏氨酸激酶,在不同物种间高度保守[3]。在正常组织中,MELK仅在睾丸中表达,其在胸腺和小肠中的表达水平非常低[4]。MELK最初被鉴定为信号转导因子,作为细胞内信号调节物发挥重要作用,并影响各种细胞和生物学过程,包括细胞周期、细胞增殖、凋亡、剪接体组装、基因表达、胚胎发育、造血和肿瘤发生等[3]。MELK可被不同外源性刺激激活,包括H2O2、肿瘤坏死因子-α(TNF-α)、硫精氨酸、离子霉素、TGF-β1、5-氟尿嘧啶(5FU)和阿霉素(DOX)等,它们可触发ASK1,TGF-β和p53信号通路[5]。此外,研究证实MELK具有广泛的底物特异性。可通过磷酸化激活MAP3K5/ASK1,以激活细胞凋亡,也可通过介导CDC25B的磷酸化调节细胞周期[6],从而导致细胞增殖和致癌。虽然MELK在其他肿瘤中已有相关研究[7-8],但在NSCLC预后判断中的作用尚未明晰。本研究基于TCGA和GEO数据库等公开的基因表达谱数据,旨在阐明MELK在NSCLC中的预后价值。
1 材料和方法
1.1数据检索方法 以“肺癌”为关键词检索肿瘤基因组图谱计划(TCGA)和基因表达谱(GEO数据库),并制定检索策略,检索流程见图1。研究类型设置为“expression profiling by array”,条目类型设置为“datasets”。所有入选数据集的样本量≥50。数据集必须包括必要信息,如生存信息、人口学特征、肿瘤分期和治疗信息等。数据库检索由两名研究人员独立进行,并对有异议的数据集进行讨论并最终达成共识。
1.2数据采集与处理 由两名研究人员独立提取所有纳入数据集中的信息。对于入选的芯片及测序数据,采用基于R软件(3.5.0版本)的循环算法计算每个数据集中基因的HR值,筛选具有统计学显著性的基因及其HR值,各个结果数据依据基因名进行匹配。根据HR值计算得出2个指标:HRh和HRl,其中HRh定义为数据集中所有大于1.0的HR值总数;HRl定义为数据集中所有小于1.0的HR值总数。分析流程见图2。

图1 文献检索流程
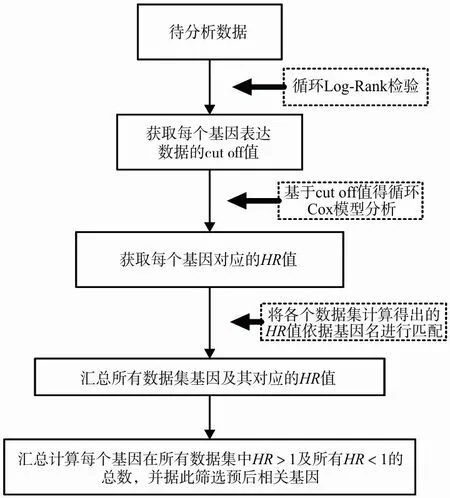
图2 基因筛选分析流程
1.3统计学分析 使用R3.5.0(R Foundation for Statistical Computing,Vienna,Austria)和GraphPad Prism 5.0软件包(GraphPad Software,Inc.,San Diego,USA)进行统计分析。两组或两组以上均值的比较采用t检验和单因素方差分析。生存曲线采用Kaplan-Meier法并进行Log-Rank检验。用单因素和多因素Cox模型计算MELK的HR值及其95%CI以评价其对预后的影响。通过CochranQ和I2检验评价基于MELKmRNA水平的各个数据集结果的异质性。当I2>50%或P<0.1时采用随机效应模型(Dersimonian-Laird法),其他采用固定效应模型(Mante-Haenszel法)。
2 结果
2.1研究特征 通过关键词检索从GEO数据库中初筛选出779个潜在相关数据集。通过样本大小和组织类型筛选后,共获取680个数据集。根据数据摘要和临床参数,最终从GEO数据库中获得7个相关研究(GSE14814[9]、GSE30219[10]、 GSE37745[11]、GSE42127[12]、GSE50081[13]、 GSE68465[14]、 GSE68571[15]),包括TCGA数据库中独立的肺腺癌、肺鳞癌数据,最后纳入8个腺癌和6个鳞癌数据集,包括1 536例肺腺癌和739例肺鳞癌患者。所有纳入数据集的基线特征见表1。

表1 纳入数据集的基线特征
注:LUAD,肺腺癌; LUSC,肺鳞癌; NR,未报道; OS,总生存期。
2.2预后相关基因筛选 基于循环算法,计算选定数据集中所有基因的HR值,并计算HRh和HRl指标。在LUAD数据集中,排列前5位且提示预后良好的基因为CAT、FRZB、CTSH、PBXIP1、NPC2,提示预后不良的基因为TK1、INPP4B、TTK、MELK、NPAS2。在LUSC数据集中,排列前5位且提示预后良好的基因分别为ARMCX6、C3orf58、CYB5R2、DGKA、DUSP9,提示预后不良的前5位基因是ABCC3、ALDH7A1、ERBB2、FTO、HTR1D。见表2、3。此外,考虑基因在不同组织中的表达水平,TK1、TTK和MELKmRNA在肿瘤组织中的表达水平明显高于正常组织,而INPP4B、NPAS2在肿瘤组织中的表达水平与正常组织相比,差异无统计学意义。
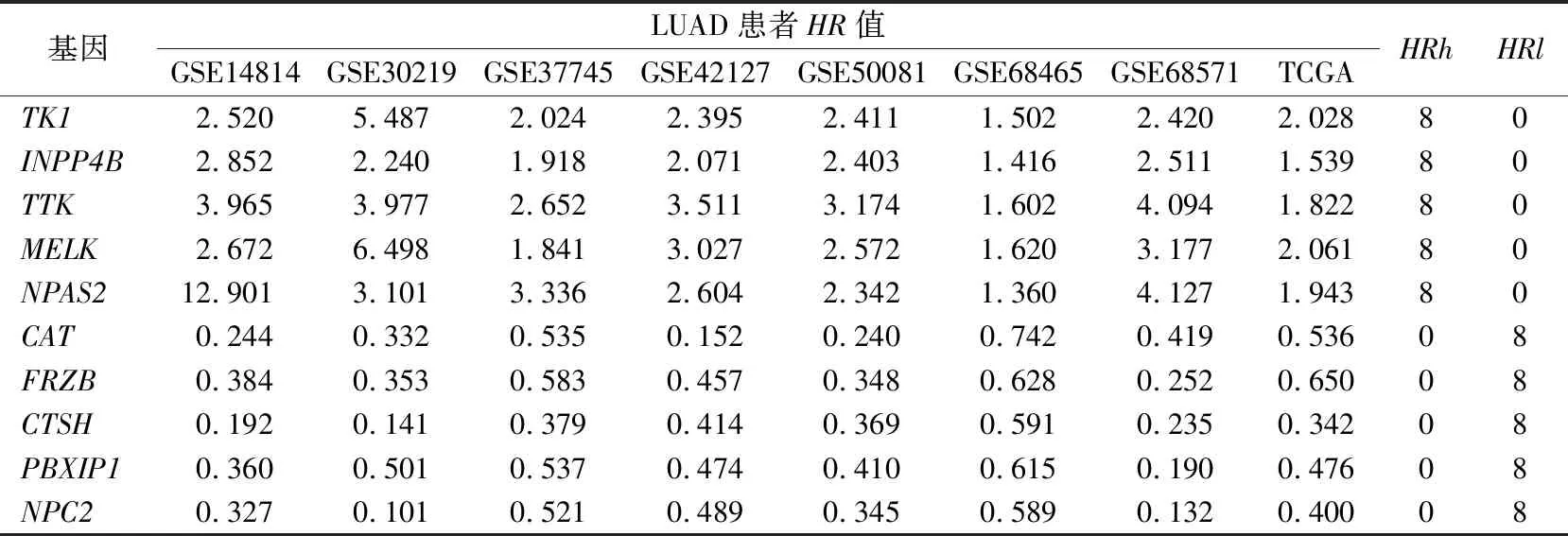
表2 LUAD患者预后相关基因的筛选

表3 LUSC患者预后相关基因的筛选
2.3MELKmRNA在肿瘤、正常组织及不同病理分期中的表达 在TCGA LUAD数据中,MELKmRNA在肿瘤组织中的表达水平显著高于正常组织(t=24.906,P<0.001,图3A),且从Ⅰ~Ⅳ期逐渐升高(F=3.779,P=0.011,图3B)。与LUAD结果相似,在LUSC数据中MELKmRNA在肿瘤组织中的表达水平亦明显高于正常组织(t=26.401,P<0.001,图3C),且从Ⅰ~Ⅳ期逐渐升高(F=4.195,P=0.002,图3D)。

注:A,正常对照及肺腺癌组织中MELKmRNA表达; B,肺腺癌不同分期MELKmRNA表达; C,正常对照及肺鳞癌组织中MELKmRNA表达; D,肺鳞癌不同分期MELKmRNA表达
图3 不同肿瘤组织及不同病理分期的MELKmRNA表达水平
2.4MELKmRNA表达水平与NSCLC患者预后的关系 对NSCLC各数据集分别进行单因素和多因素Cox模型分析。LUAD和LUSC患者各数据集中MELKmRNA的P值、HR值和95%CI值见表4和表5,LUAD和LUSC患者各数据集的生存曲线见图4和图5。
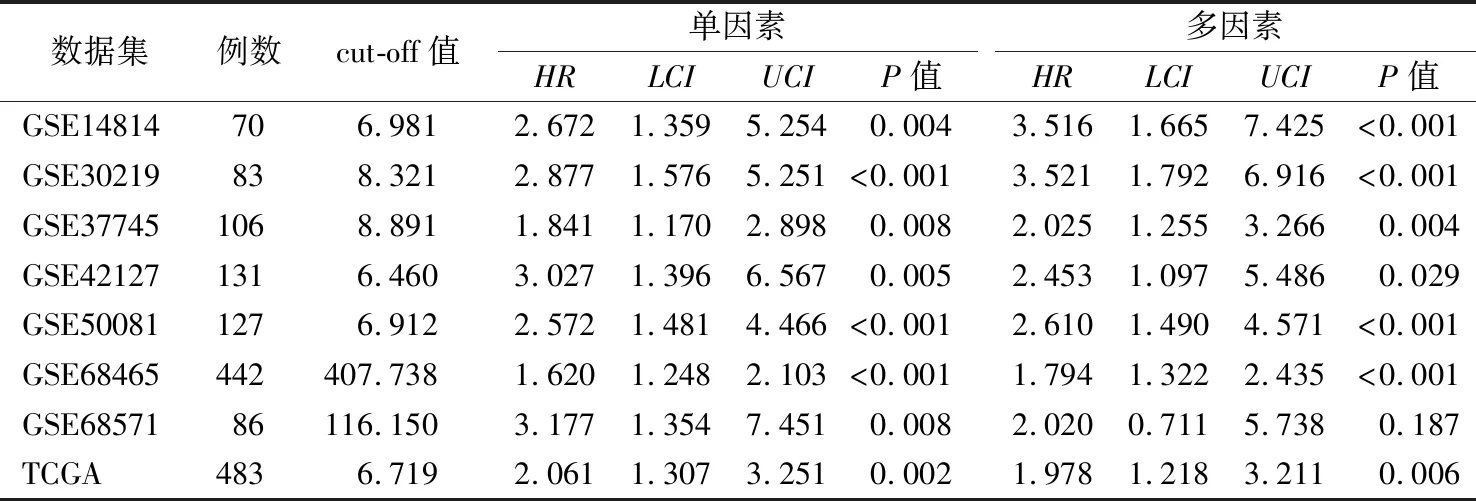
表4 肺腺癌数据集Cox模型分析结果

表5 肺鳞癌数据集Cox模型分析结果

注:A,GSE30219;B,GSE37745;C,GSE42127;D,GSE50081;E,GSE68465;F,GSE14814;G,GSE68571;H,TCGA。
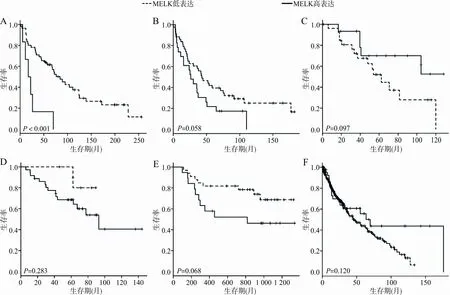
注:A,GSE30219;B,GSE37745;C,GSE42127;D,GSE50081;E,GSE14814;F,TCGA。
2.5对LUAD和LUSC患者进行MELK影响预后的联合分析 对LUAD的Cox多因素生存分析结果效应量进行meta合并,异质性检验显示其无明显的统计学异质性(I2=0.0%,P=0.562),故而采用固定效应模型合并HR值。结果显示,MELKmRNA高表达与较低的总生存率(OS)显著相关(合并HR=2.162,95%CI:1.796~2.604),结果见图6A。对LUSC的Cox多因素生存分析结果效应量进行meta合并,异质性检验显示存在统计学异质性(I2=67.1%,P=0.009),故采用随机效应模型合并HR值。结果显示LUSC患者MELKmRNA和OS之间无显著关联(合并HR=1.304;95%CI:0.953~1.786),结果见图6B。

图6 肺腺癌和肺鳞癌患者多因素生存分析的森林图
3 讨论
近年来NSCLC发病率和死亡率在各类肿瘤中均居于前列,严重威胁着人类健康。因此,寻找新的生物标志物对NSCLC的诊断和治疗具有重要意义。目前同类研究较少,Piao等[16]基于GEO数据利用GEO2R功能寻找差异表达基因,并通过STRING、Cytoscape和MCODE构建蛋白质相互作用网络(PPI),以筛选关键基因。该方法主要基于基因表达差异,而本研究所采用的方法主要基于预后价值。与上述网站检索方法相比,本研究方法存在以下几点差异:首先,本研究基于每个独立的数据集进行分析,是基于较少的样本量得出的结果,合并后更加稳健。通常在科研设计中,组间期望差异越小,所需总样本量越大,反之,样本量越大,越易得到显著的结果,同时也意味着较高的假阳性率。其次,HRh和HRl可用来判断其相应基因的预后价值。结合HRh和HRl具体数值有助于明确基因的预后价值,即HRh值越高,同时HRl值越低,表明该基因为NSCLC患者的高风险基因;呈相反趋势则表明该基因对NSCLC患者的保护作用越强,因此保证了筛选结果的准确性。Li等[17]采用WGCNA分析法鉴定肺腺癌中与生存相关的表达模块,其从识别的模块中,获取了3个关键基因UBE2C,TPX2和MELK,这一过程主要是通过生物信息方法并基于单个数据集来实现的。
在本研究中,LUAD和LUSC患者肿瘤组织中MELKmRNA表达水平明显高于正常组织,且从Ⅰ~Ⅳ期逐渐升高。有研究显示,MELK在多种癌症中表达升高,包括乳腺癌、肝癌、胃癌、急性髓系白血病、胶质母细胞瘤、肾癌等[7,18-20]。此外,MELKmRNA的高表达与LUAD患者预后不良相关,这与Speers等[21]和Kuner等[8]在乳腺癌和前列腺癌患者中的研究结果相似。本研究中,LUSC数据集的总HR值显示无统计学意义,但它仍然揭示了一种趋势,即MELK高表达与LUSC患者的不良预后可能相关。这种预后差异可能归因于肺腺癌及鳞癌不同的组织学特征、不同的治疗手段以及对治疗手段的反应率的差异造成的。本研究是基于公共数据的挖掘研究,由于不同数据产生于不同的检测平台,包括基因芯片和测序数据,数据存在一定的异质性,本研究的结果还有待更高级别证据的研究进一步证实。综上所述,MELKmRNA在肺腺癌中的表达水平越高其预后越差,MELK有望成为肺腺癌患者治疗的潜在的分子靶点。
