1例假性甲状旁腺功能减退症与GNAS基因突变
2020-06-11周希杨治芳
周希 杨治芳
南昌大学第一附属医院内分泌科 330006
假性甲状旁腺功能亢进症(PHP)是一种罕见的异质性疾病,具有家族遗传倾向,其特征是由于对靶器官中的甲状旁腺激素(PTH)的抗性而导致的低钙血症和高磷血症[1]。PHP有3种形式,即PHP-1、PHP-2和伪假甲状旁腺功能亢进症。基于Albright遗传性骨营养不良(AHO)的存在与否,PHP-1进一步分为3种不同亚型(1a、1b和1c),PHP-1a和PHP-1c都显示AHO的功能,但PHP-1b没有,PHP-1a与PHP-1c的区别在于它含有编码鸟嘌呤核苷酸结合蛋白(Gsα)的基因中的失活突变。本文通过临床表现及遗传学分析,报道1例PHP家系。
1 病例介绍
病例1,患者为女性,21岁,因无月经来潮就诊于外院,诊断“甲状腺功能减退症”,给予左甲状腺素钠片(25 μg qd),症状未见好转,遂就诊于南昌大学第一附属医院门诊。患者家属诉患者8岁时有过四肢抽搐现象,与癫痫发作症状相似。既往史:否认类固醇激素使用史。个人史:足月顺产。查体:身高126 cm,体重41.5 kg,体重指数:26.14 kg/m2。反应稍迟钝、脸圆、颈短、甲状腺未触及肿大、双肺呼吸音清、未闻及干湿性啰音。心律齐,各瓣膜听诊区未闻及病理性杂音。腹平软,无压痛、反跳痛,肝脾肋下未触及。乳腺可见发育,腋毛缺失,阴毛缺如,大阴唇见幼儿外观,双手伸开时见两掌骨第Ⅳ掌骨远端凹陷(图1)。实验室检查:患者血生化及相关激素水平见表1。影像学检查:X线示:左手三角骨、豆状骨、钩状骨形态不规则,骨质密度不均匀增加,边缘模糊(图2)。妇科B超:子宫大小正常,左侧卵巢大小2.6 cm×2.2 cm×1.8 cm,右侧卵巢大小2.6 cm×2.0 cm×1.3 cm。染色体:46,XX。

表1 病例1及病例2相关化验结果
注:PTH:甲状旁腺激素;Ca:钙;P:磷;LH2:血清促黄体生成素;FSH:卵泡刺激素;E2:雌二醇;TSTO:睾酮;sTSH:高敏促甲状腺激素;Anti-TG:抗甲状腺球蛋白抗体;Anti-TPO:抗甲状腺过氧化物酶抗体
病例2,患者为男性,11岁,为病例1的弟弟,因生长迟缓入院。既往史:否认类固醇激素使用史。个人史:足月顺产。查体:身高129 cm,体重:32 kg,体重指数:19.2 kg/m2,智力正常、视力差、脸圆、颈短、甲状腺未触及肿大、双肺呼吸音清、未闻及干湿性啰音。心律齐,各瓣膜听诊区未闻及病理性杂音。腹平软,无压痛、反跳痛,肝脾肋下未触及。外生殖器:无阴毛,睾丸容积:左侧4 cm,右侧4 cm,质软,未触及肿物,阴囊未见色素沉着,附睾未触及肿大。握拳时见两掌骨第Ⅳ掌骨远端凹陷(图3)。实验室检查:患者血生化及相关激素水平详见表1。影像学检查:X线示:左手第4掌骨短小,拟为先天性发育异常(图4)。头颅MRI:双侧额叶及基底节区对称性高密度影,结合病史,拟为转移性钙化灶(图5)。甲状腺彩色超声:甲状腺弥漫性病变;双侧叶甲状腺结节。考虑甲状腺结节可能(T1-RADS 3类)。
两患者父亲:身高160 cm,智力正常,查体未见异常。两患者母亲:身高136 cm,脸圆,孕5产2,家族遗传系谱见图6。
检测病例2的鸟嘌呤核苷酸结合蛋白刺激多肽(GNAS)基因发现c.2522delT(编码区2522号核苷酸T缺失),该变异导致从第842位Arg开始的氨基酸合成发生改变,并在改变后第5个氨基酸终止(p.Arg842AlafsTer5)。同样,病例1及其母亲GNAS基因8号外显子密码子2522上都缺失一个碱基T,均造成移码突变(p.Arg842AlafsTer5),而病例1父亲检测结果无异常(图7)。
根据病例1及病例2的典型临床表现:(1)脸圆,体态矮胖,发育异常,智力发育迟缓,对称性第4掌骨缩短。(2)实验室检查低钙、高磷血症、高PTH。(3)影像学特征。(4)基因突变系列分析结果。诊断为PHP。
由于目前并无针对病因治疗的具体措施,主要是控制症状,使血钙接近正常或正常,减少并发症的发生。故给予补钙及左甲状腺素治疗,2个月后随访结果见表2。

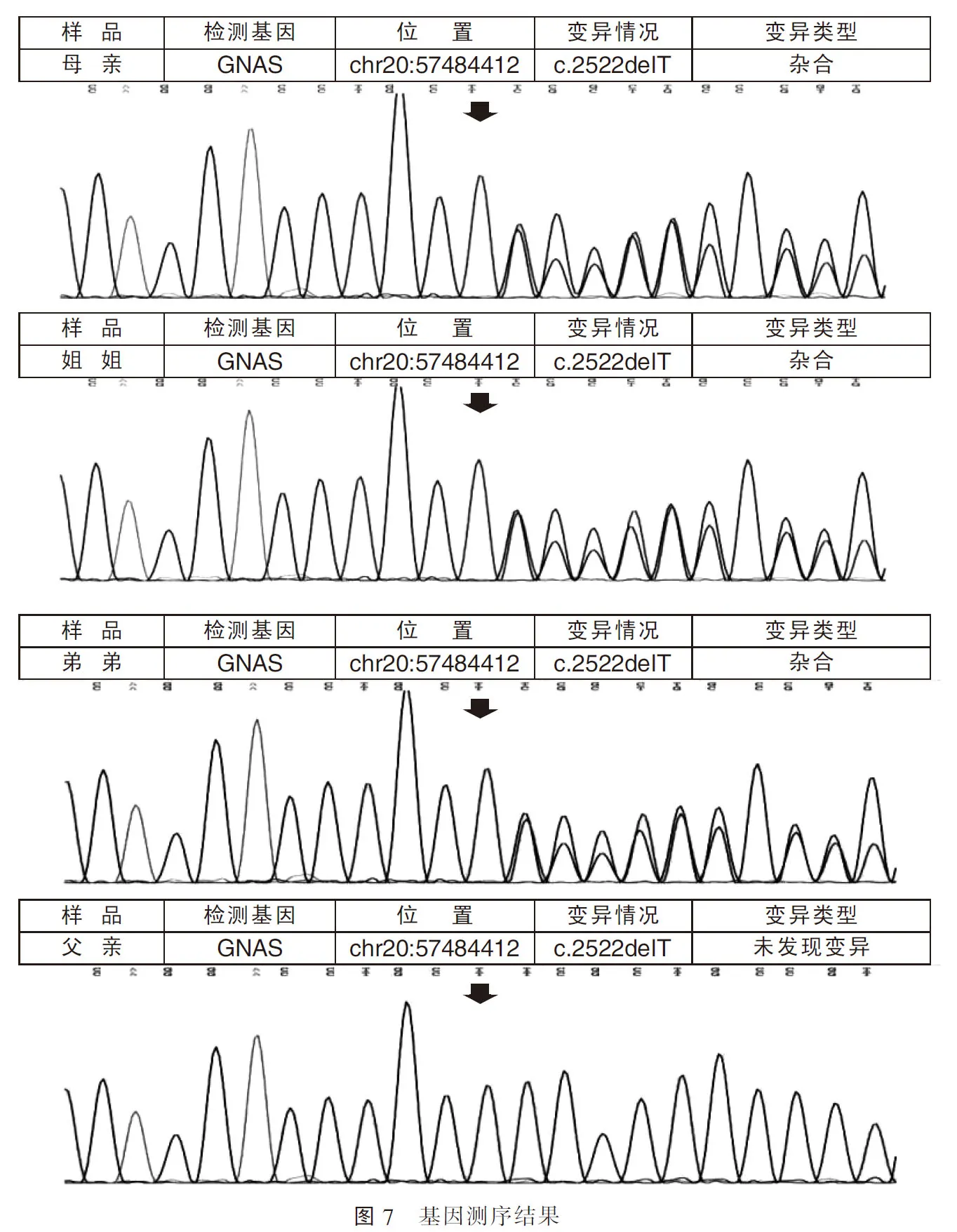
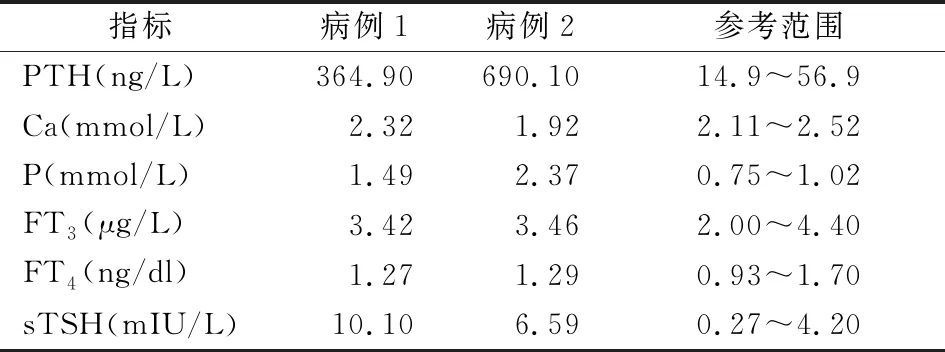
表2 病例1及病例2在2个月后随访的结果
注:PTH:甲状旁腺激素;Ca:钙;P:磷;sTSH:高敏促甲状腺激素
2 讨论
PHP是第一种激素抵抗综合征,其特征在于低钙血症、高磷血症、PTH水平升高和AHO。AHO的临床特征是肥胖、圆脸、身材矮小、短指、皮下骨化和智力低下。PHP-1a的特征是靶器官对PTH的抵抗和AHO的特征,而PHP-1b经典表现为激素抵抗仅限于没有AHO征象的PTH[2]。PHP-1a由GNAS基因显性突变引起,GNAS是染色体20q13.3上复杂印迹基因座的一部分,Gsα由GNAS的外显子1~13编码。GNAS突变的母系遗传导致PHP-1a,而相同突变的父系遗传导致PPHP,其中可见AHO的特征但不存在激素抗性[3]。
本文病例1及病例2均有明显的AHO特征,且母系遗传特点明显,而其父亲未见病变,符合PHP-1a型。Hanna等[4]研究提出,早发性肥胖是GNAS改变的特征,表明Gsα降低表达是一个促成因素,而生长板(可能还有其他组织)中的Gsα单倍体不足,可能是PHP-1a患者出生时轻度生长受限的原因。病例1及病例2均存在TSH、甲状腺抗体水平升高,病例2甲状腺彩色超声示甲状腺弥漫性病变,考虑存在甲状腺自身免疫。在PHP-1a、PHP-1c中,除PTH抗性外,甲状腺功能减退症、生长激素缺乏症和性腺功能减退症也可反映靶器官对TSH、生长激素释放激素和促性腺激素的抵抗,考虑可能与有共同Gsα耦联的受体有关[5-7]。研究显示,亚临床甲状腺功能减退症是最早提示PHP-1a的特征之一,并且存在于70%的PHP-1a病例中[8]。Femandez-Rebollo等[9]表明,大多数PHP-1a患者具有抗性,通常是轻微的,并且在儿童期或青春期表现出来,通常缺乏甲状腺肿和甲状腺自身抗体。Balavoine等[10]研究发现,PHP患者中存在甲状腺过氧化物酶抗体及甲状腺球蛋白抗体。既往有研究提出,甲状腺自身抗体存在于10%~20%的女性中,并且TSH增加抗体自身免疫性[11]。尚未阐明PHP-1a导致甲状腺功能减退症的机制。治疗是给予左甲状腺素以达到正常的TSH水平。病例1因无月经来潮就诊,既往有低钙抽搐病史,诊断为“甲状腺功能减退症”,并补充左甲状腺素,检查示患者LH2偏高值,雌激素低下。性腺功能减退症表现在PHP-1a患者中也很常见,这些患者存在青春期延迟或性成熟不全,女性患者通常出现月经异常,如原发性闭经/月经稀发,有时还有不孕症[12]。因此,PHP-1a可能与甲状腺功能减退症混淆,因为这两种疾病都可表现为身材矮小、肥胖和月经紊乱。另外,也应与癫痫、矮小症相鉴别。本研究通过GNAS基因检查,发现GNAS基因外显子8中碱基缺失c.2522delT(编码区2522号核苷酸T缺失),造成移码突变,导致突变等位基因中的过早终止密码子,从而产生截短的非功能性蛋白质。该变异的致病性尚未见文献报道,为一新的突变。迄今为止,已报道超过400种失活的GNAS突变,包括移码、错义、无义、剪接位点突变、框内缺失或插入,以及全部或部分基因缺失[13]。描述的所有突变中约20%的热点突变在外显子7中[14]。既往有过类似报道,Rickard和Wilson[15]曾对1例AHO患者研究发现,其内含子7剪接受体位点的突变,导致外显子8单个G核苷酸(c.586delG)的缺失、移码,使得外显子8中的过早蛋白质截短。
PHP发病罕见,危害大,获得正确的分子诊断也可能是困难且耗时的。对患者状态进行分析有助于规划适当的心理测试和制定随后的特定治疗。此外,因PHP是常染色体遗传性疾病,对于已经确诊为PHP的患者在孕育第3代时提前进行受精卵筛查,对孕育健康下一代有非常重要的意义。
