基于富集放大反应的双点电位滴定法测定谷物中氟含量
2020-06-11刘晓庚吴李瑞董思佳杨奕明刘季敏胡妍君张宇豪秦怡航
刘晓庚 吴李瑞 董思佳 杨奕明 刘季敏 胡妍君 张宇豪 秦怡航
(南京财经大学食品科学与工程学院;江苏省现代粮食流通与安全协同创新中心; 江苏高校粮油质量安全控制及深加工重点实验室1,南京 210023) (安徽国星生物化学有限公司2,马鞍山 243100)
氟既是人体必需微量元素,但又是一种累积性物质,体内含量超过一定程度会引起氟中毒。氟以氟离子形态广泛存在于水体、土壤、动植物及食物中。目前测定食物中氟的方法主要有离子选择电极电位滴定法(GB/T5009.18—2003)[1-5]、极谱法[6]、分光光度法[7,8]、荧光分析法[9]、气相色谱法[10]、高效液相色谱法[11]和离子色谱法[12]等。电位滴定存在检测限较高、极谱法有重现性不佳、光度法和荧光法有试剂不易得到和稳定性差、而色谱法又存在操作较繁琐和测定成本高等不足[13]。人们对生态环境和食品安全的要求越来越高,为寻得既快速准确经济且适合基层检测的谷物微量氟测定新方法,在大量调研和测定方法的探究性实验基础上,探究出高倍富集放大双点电位滴定法测定微量氟的新方法,并获得了较理想的检测效果。
1 高倍富集放大反应测定氟的原理
高倍富集放大反应测定法的基本原理是在一定条件下,Ca2+、F-与特定试剂组作用,定量生成是其氟含量5倍的具有较弱极性的难溶化合物氟磷酸钙[Ca10(PO4)6F2,(Ksp(Ca10(PO4)6F2) = 2.48×10-6)][14,15],从而使氟富集并分离,在酸性环境下将氟磷酸钙完全溶解,以EGTA为滴定剂,用双电点电位滴定法[16]测其钙含量[17],转换成试样中氟含量。其相关过程的反应式为:
①10Ca2++2F-+6(PO4)3-==Ca10(PO4)6F2↓
②Ca10(PO4)6F2+2HCl==10Ca2++Cl2-+2HF+6(PO4)3-
③Ca2++H2Y2-==CaY2-+2H+
其计算式为:

式中:Ve为滴定液体积/mL;Ve,0为样品空白滴定体积/mL;cEGTA为EGTA标准液浓度/mol/L;ms为试样质量/g;MF为氟的摩尔质量/g/mol;k为干物质含量。
2 材料与方法
2.1 材料与试剂
小麦、玉米、稻谷等由南京财经大学中加生态储藏中心提供。
EGTA、NaF、CaCl2、NaCl、KOH、Na3PO4、NH4Cl、HCl、HNO3、HAc、明胶和过氧化钠等均为分析纯试剂。实验用水为去离子纯净水。
总离子强度调节缓冲剂(TISAB)由NaCl 1.0 mol/L、HAc 0.25 mol/L、NaAc 0.75 mol/L、柠檬酸钠0.05 mol/L组成,pH 5.0,总离子强度为2.75。
2.2 仪器与设备
ZDJ-4A型自动电位滴定仪;Ca离子选择性电极;E-201-C pH复合电极。
2.3 方法
2.3.1 样品前处理
参照粟智[19]的氧弹燃烧法进行预处理,即:将干燥测定水分(<8%)的试样磨碎,过60目筛,称取试样5.00 g,与0.25 g过氧化钠助燃剂混匀,压片。将5 mL 0.5 mol/L KOH吸收液加入氧弹器中,放置压好的试剂片,灰化,用吸收液洗净弹壁,合并后用0.2 mol/L HNO3中和至pH 7,并定容至100 mL备用。
2.3.2 测定操作方法
按王洪友[15]的方法,取一定量试液于刻度试管,依次加一定量的2 mol/L Na3PO4、2 mol/L KOH、2 mol/L HAc和1 mol/L CaCl2(此4种溶液后简称为含Ca2+复合富集剂)混合均匀后,再加入明胶,定容,静置,待明胶粉末完全吸水膨胀后,于60 ℃水浴中加热,搅拌至明胶完全溶解,得均匀透明的溶胶;将此胶倒入聚乙烯杯中,室温下冷却至溶胶完全凝固;再于恒温浴和40 W超声波辅助下反应一定时间至沉淀层不再增加时,分出固体物并用35℃水洗至pH约7。再用2 mol/L HCl将固体物完全溶解,用1 mol/L KOH调至pH约7,并用TISAB定容至100 mL。取该试液20.00 mL,加1 mol/L KOH 5 mL,加钙指示剂,混匀,于自动电位滴定仪用EGTA标准液(用碳酸钙作基准物标定)滴定至终点,并记录计量点附近两组V,E数据(V1,E1;V2,E2)并用双点法计算终点体积Ve,同一样品平行测定3次。以水做空白实验Ve,0。
2.3.3 富集放大反应条件的确定2.3.3.1 单因素考察方法
影响高倍富集放大反应的因素有:富集剂用量、反应时间、反应温度、反应体系酸度(pH)和搅拌速度等。用单因素变量法考察这些因素对测定的影响(以氟含量和RSD为衡量指标),并确定其适宜条件。
2.3.3.2 优化实验方法
在单因素实验基础上,为更准确地确定其主要因素的最优条件,对富集剂用量、反应时间、反应温度、反应体系pH四个主要影响因素用L9(34)的正交实验进行优化,以确定其最适宜条件。
2.3.4 方法学指标的考察2.3.4.1 标准曲线和检测限的测定
在最适宜条件下,取氟标准液,浓度(-lgcF)分别为1.0, 2.0, 3.0, 4.0, 5.0,6.0, 7.0 mol/L,测得其双点电位滴定法E~-lgcCa标准曲线,并根据其斜率(k)和空白值测定的标准偏差(Sb)算得钙的检测限(LODCa=3Sb/k)[17],再据氟与钙的富集放大反应的计量关系便可求出本法测定氟的LODF(=LODCa/放大倍数)。
2.3.4.2 方法精密度和准确度的测定
方法的精密度以相对标准偏差(RSD)表示。方法准确度以加标实验的回收率表示。
2.3.5 实际样品的测定
在最适宜条件下,按2.3.1制得13个样液,并用2.3.2操作方法对其分别进行测定。
2.3.6 数据处理
数据采用Excel 2017和Origin9.0绘图和数据处理。
3 结果与讨论
3.1 高倍富集放大反应的选定
在单用CaCl2富集F-,但效果不佳且无放大效应后。考虑到氟磷酸钙含氟且难溶,特别是反应对氟的放大效应达10倍[14,15],实验结果见图1。0.1 mol/L含Ca2+复合富集液对微量F-有良好的富集放大效果。因此,选择含Ca2+复合液作为F-的富集放大剂。

图1 0.1 mol/L含Ca2+复合富集剂与 1.0×10-7~1.0×10-5mol/L NaF反应的富集放大效果
3.2 富集放大反应条件的单因素实验
3.2.1 富集剂用量的影响
由图1可见,当c(F-)≥1.0×10-6mol/L时富集产生沉淀层十分明显,因此固定c(F-) 1.0×10-6mol/L,仅改变含Ca2+复合富集剂浓度且分别为1.0、0.5、0.2、0.1、0.05、0.01、0.005、0.001、0.000 5、0.000 1 mol/L作实验,结果见图2a。若复合富集剂用量太少,会因体系浓度过稀而达不到沉淀条件;如果用量过多则会导致配位效应突出而使沉淀富集作用削弱。实验证明:1.00×10-6mol/LF-和0.05~0.20 mol/L含Ca2+复合富集剂反应产生的沉淀富集放大效果为佳,故选用Ca2+复合富集剂的用量为n(F-)∶n(复合富集剂)= 1∶5×104~2×105为宜。
3.2.2 富集放大反应酸度的影响
其他条件均不变,通过仅改变富集反应体系的酸度(pH)且分别为3、4、5、6、7、8、9、10、11、12下,测得其结果如图2。富集反应体系酸度控制在pH5 ~7为宜。
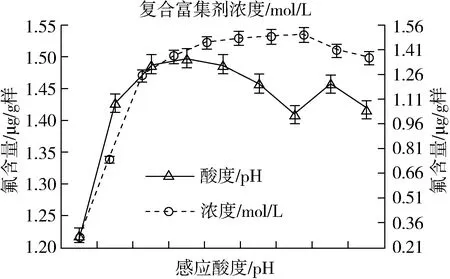
图2 反应酸度和复合富集剂对测定的影响
3.2.3 富集放大反应温度的影响
其他条件均不变,通过仅改变富集反应的温度且分别为10、15、20、25、30、35、40、45 ℃,测得其结果如图3,富集反应温度控制在15~25 ℃为宜。

图3 反应温度、反应时间和洗涤次数对测定的影响
3.2.4 洗涤次数的影响
其他条件均不变,通过仅改变对富集所得固体物的洗涤次数且分别为0、1、2、3、4、5、6次下(洗涤的用水量相同),测得其结果见图3。沉淀物的洗涤对测定有显著影响,特别是当洗涤次数少于3次时,不仅测定结果严重偏高,而且测定的RSD大,重现性、稳定性和准确性均差,达不到分析要求;当洗涤次数达4次以上时,其回收率趋近100%和RSD小且稳定,完全符合微量分析要求;而洗涤次数太多不仅浪费而且会因过度洗涤使沉淀物流失而导致测定结果偏低。因此洗涤次数以4次为宜。
3.2.5 富集放大反应时间的影响
其他条件均不变,通过仅改变富集反应的时间且分别为30、60、90、120、150、180、210、240、300 h下,测得其结果见图3。富集反应时间应控制在180 min以上为宜。
3.2.6 搅拌速率对富集放大反应的影响
其他条件均不变,通过仅改变富集反应的搅拌速率且分别为0、10、30、60、100、150、200、250、300、400 r/min下,测得其结果见图3。搅拌速率对测定的影响不十分显著。但搅拌过慢,反应充分,测量结果偏低,搅拌快,会产生气泡形成大漩涡而富集不彻底,因此富集过程中搅拌速率在200~300 r/min为宜。
3.3 正交优化实验
为获得此富集放大反应的最适条件,根据单因素实验结果,对富集剂用量、反应时间、反应温度、反应体系pH四个主要影响因素采用L9(34)正交优化实验,其实验设计与结果见表1,结果分析见表2。
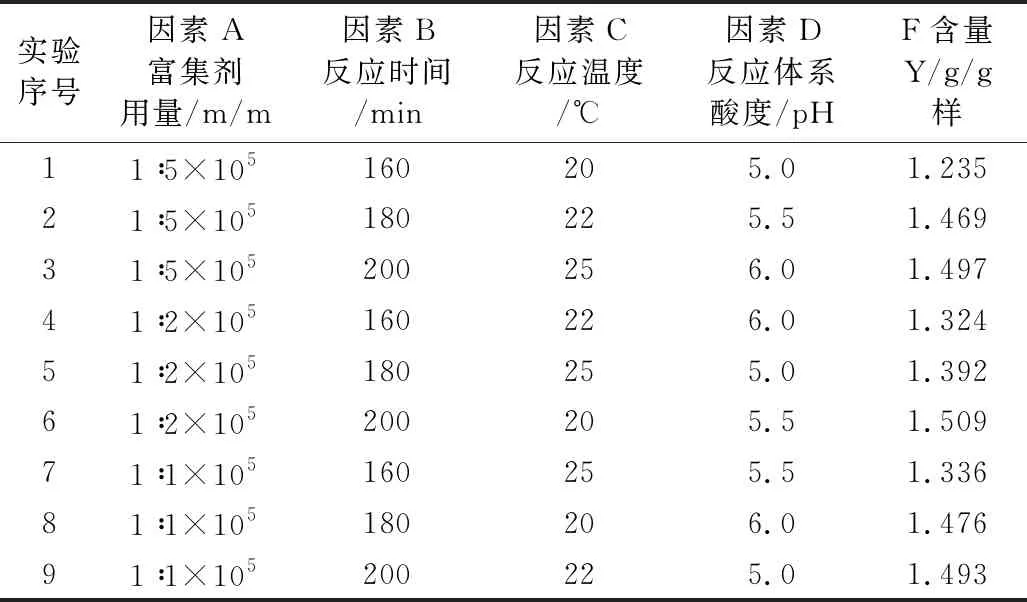
表1 正交实验设计及结果 (n=3)
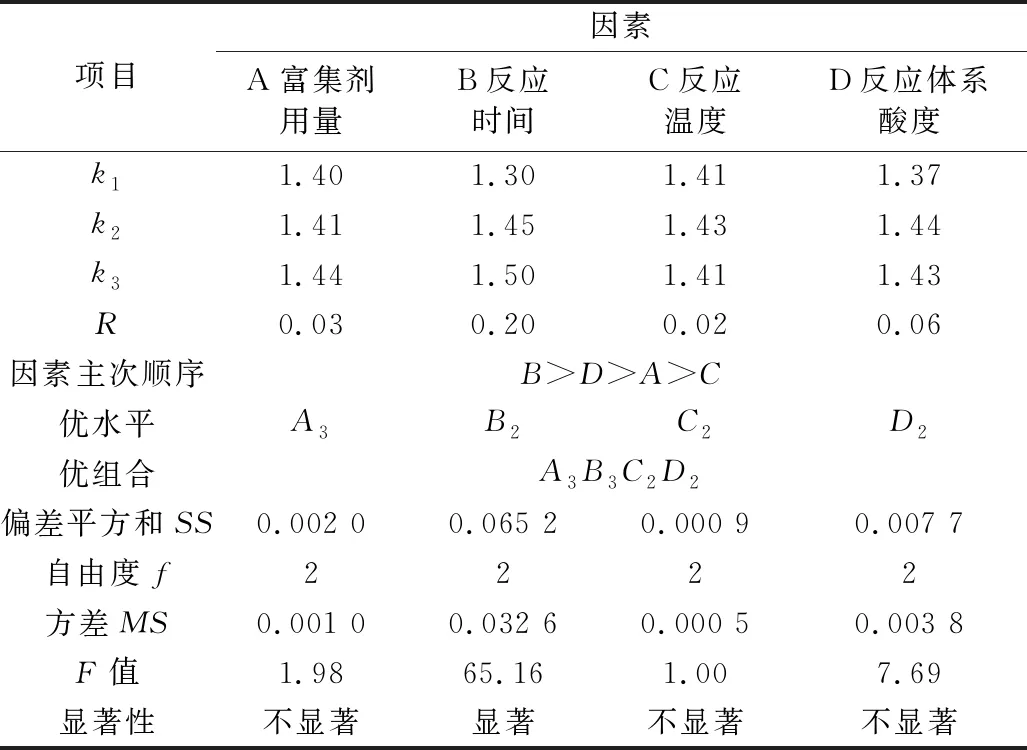
表2 正交实验结果的分析
注:F0.05(2,2)=19.0,F0.01(2,2)=99.0。
从表2可见,四个被优化的主要因素对测定的影响程度是:B(富集反应时间)>D(反应体系酸度)>A(复合富集剂用量)>C(反应温度);除富集反应时间影响显著外,其他影响因素均不显著(P<0.05),且反应的温度对测定的影响则最小,原因是所考察的反应时间水平间跨度较大,而其它考察因素水平间相关甚小。正交优化所得最适宜条件为A3B3C2D2,即富集反应时间200 min,反应温度22 ℃,反应体系酸度pH 5.5,复合富集剂用量为(m氟/m复合剂) = 1∶1×105。
3.4 标准曲线和最低检测限的测定
在最适宜条件下按2.3.4.1的方法测得其E~-lgcCa标准曲线见图4。从图4可见,cF在1.0×10-6~1.0×10-1mol/L范围内均有良好定量的函数关系:E=38.657 lgc-174.44 (R2=0.996)。并可求得双点法测钙的检测限(LODCa=2.8×10-6mol/L)。再依据富集放大反应中钙与氟的计量关系,便可得到本法测定氟的检测限LODF=5.3×10-3μg/g,与高灵敏的极谱法[20]的检测限相当。
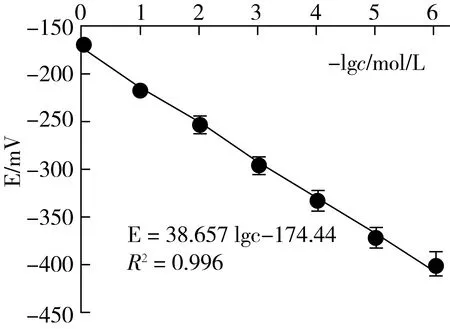
图4 双点电位滴定钙离子的E~ (-lgcCa)标准曲线
3.5 精密度与准确度实验的测定
在最适宜条件下按2.3.4.2的方法分别测得稻米和米皮中氟含量的RSD和加标实验的回收率及其RSD,结果见表3。由表3知,测得含氟量相差甚大的稻米和受氟污染米皮的RSD为1.9%~3.9%和加标回收率为91.5%~100.3%,且氟含量低于200 μg/g样的测定效果更佳。可见本法测氟的重现性和精确性均良好。
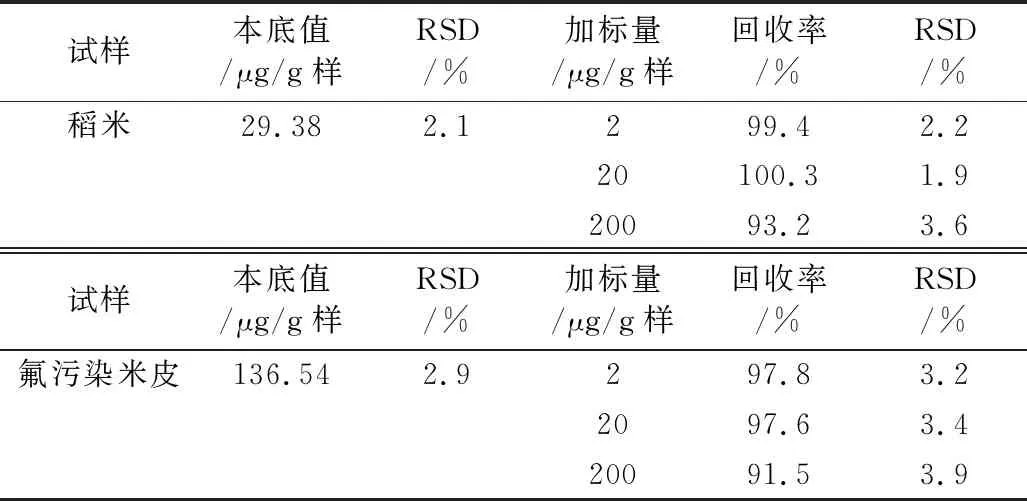
表3 本法精密度和准确度实验结果 (n=3)
3.6 干扰因素实验
谷物中存在的Mg2+和Fe3+会影响本法测定[15,17]。因此测量时采取的排除干扰措施是:①在复合富集剂和TISAB中均含对Mg2+和Fe3+有掩蔽作用的柠檬酸或其盐。②双点电位滴定法的滴定剂是对Ca2+选择性强的EGTA而不是选择弱的EDTA,因为在EGTA滴定Ca2+条件下Mg2+和Fe3+已转化为难溶的氢氧化合物故不影响此测定[21]。可见本法测定均已设计了消除干扰的处理无需额外的相应措施。
3.7 实样的测定
按2.3.1方法处理试样,再按2.3.2在最适宜条件下进行测定,结果见表4。由表4可知,原粮中含氟量为1.27~1.48 μg/g样,加工副产物中含氟量为1.84 ~ 2.67 μg/g样,加工制品的小麦粉和稻米中含氟量分别为0.63 μg/g样和0.57 μg/g样;试样本底和加标的测定的RSD分别为1.9%~2.7%、0.8%~2.3%,均比较稳定,但本底值的RSD比加标的稍大,且不同测定对象RSD有差异(这可能是测定对象的组成成分存在明显差异所致)。但实验结果是完全符合微量分析的要求。
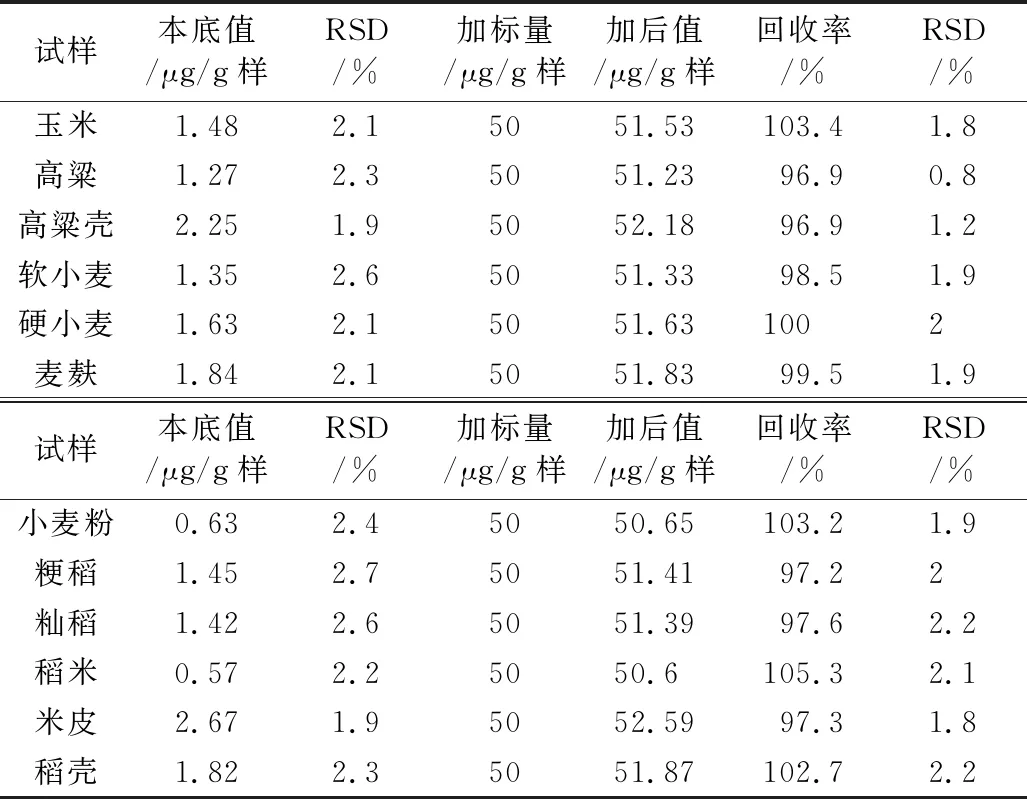
表4 实样及其回收率测定结果(n=3)
4 结论
通过富集放大反应和双点电位滴定相结合的方法,可以快速、准确地测定谷物中微量至常量的氟。对谷物中氟的富集、测定方法与条件进行了验证分析,对影响富集反应的主要参数进行了优化,所得最适条件是:含Ca2+复合富集剂用量为(m氟/m复合剂)=1∶1×105,富集反应200 min,反应温度22 ℃,反应体系酸度pH 5.5,搅拌速率200~300 r/min,沉淀物洗涤4次。该法的LODF=5.3×10-3μg/g样,当cF在1.0×10-6mol/L~1.0×10-1mol/L内呈良好定量关系:E= 38.657 lgc-174.44 (R2=0.996);且加标回收率为91.5%~100.3%,测定结果较稳定(RSD≤2.7%)。本法集合了富集反应的高倍放大效果和双点电位滴定法高精准的特点,测定选择性高,Mg2+、Fe3+干扰小,测定浓度范围大,测定速度快,灵敏度较高,可作为微量氟的定量分析方法,为修订GB/T5009.18-2003《食品中氟的测定》提供参考。
