三种跳虫肠道菌群的多样性分析及功能预测
2020-06-10马欣然渠凤甜谢桂林赵红庆
陈 伟, 陈 霞, 李 娟, 马欣然, 崔 薇, 渠凤甜, 谢桂林,*, 赵红庆,*
(1. 东北农业大学生命科学学院, 哈尔滨 150036; 2. 中国疾病预防控制中心传染病预防控制所, 北京 102206)
弹尾纲(Collembola)动物,俗称跳虫(springtails),隶属于节肢动物门(Arthropoda)六足总纲(Hexapoda),是一类原生无翅而有附肢的内口式低等六足动物,其种类繁多且分布广泛,是陆地生态中最成功的土壤动物类群之一(高艳, 2007)。跳虫主要以土壤中腐败的动植物残骸、腐殖质、细菌和真菌为食(陈建秀等, 2007)。在土壤生态系统中它们有助于有机物的分解和养分流通,同时能抑制植物病虫害,不同程度地同化化学污染物,在土壤改良、物种多样性维护及生态毒理学监测等方面发挥着重要的作用(Rusek, 1998)。
跳虫肠道是一个对陆地环境微生物具有筛选性的定植场所,其形态为简易的杆状,分前肠、中肠和后肠。前肠和后肠的平均体积分别为0.21和0.06 nL,中肠的体积会随摄食活动而有所改变(0.7~11.2 nL)(Thimmetal., 1998)。Buller(1909)在跳虫消化道内发现真菌孢子,首次证明跳虫肠道内有微生物存活。Visser等(1987)用麦芽提取物琼脂平板从Onychiurussubtenuis的粪便中培养出拟青霉属Paecilomyces和枝孢霉属Cladosporium等真菌。Borkott和Insam(1990)用几丁质琼脂平板从白符跳Folsomiacandida的粪便中分离出能降解几丁质的嗜麦芽黄单胞菌Xanthomonasmaltophilia和短小杆菌属Curtobacteriumsp.。Thimm等(1998)用扫描电子显微镜观察到白符跳F.candida的中肠和后肠中存在大量的细菌,并采用YT琼脂平板分离出一种丝状真菌薄状枝顶孢霉菌Acremoniumcharticola, 以及植物梨火疫病菌Erwiniaamylovora, 成团泛菌Pantoeaagglomerans和头状葡萄球菌Staphylococcuscapitis等11种细菌。Hoffmann等(1999)研究跳虫摄食携带荧光素酶标记基因的细菌后的消化情况,结果证明摄食活动能改变肠道菌群的结构(Hoffmannetal., 1999)。
近十几年来,随着现代分子生物技术的发展,跳虫身上定植的大量微生物已被鉴定出来。其中Czarnetzki和Tebbe(2004)通过16S rDNA扩增和单链构象多态性(single strand conformation polymorphism, SSCP)分析,揭示了8种跳虫的菌群多样性。Agamennone等(2015)采用16S rDNA扩增子测序分析了实验室饲养的柏林白符跳F.candida与野生型赞丹F.candida的细菌菌群结构差异性。Anslan等(2016)使用高通量测序技术研究了3种跳虫肠道及其附肢的真菌菌群随季节的变化。在跳虫菌群功能研究方面,Agamennone等(2018)研究了从5种跳虫肠道内分离的46种菌株的抗菌性特点。王立秀等(2018)研究了从阿南原等跳Proisotomaananevae肠道内分离的20种菌株产纤维素酶的特点。Agamennone等(2019)通过宏基因组测序分析揭示了白符跳F.candida细菌菌群与抗菌活性和碳水化合物代谢相关基因的多样性。总体而言,关于跳虫肠道菌研究的报道较少,所涉及的跳虫种类也非常有限,绝大多数跳虫肠道菌群的多样性是未知的,对它们的菌群功能也是了解较少。
在本研究中,我们采用16S rDNA扩增子测序法对分别采集自我国3个地区的3种跳虫的肠道菌群进行分析,旨在揭示其肠道菌群的多样性及相关功能,为进一步有效地从跳虫肠道内容物中选择性分离可培养菌和探索菌群的相关功能提供理论依据。
1 材料与方法
1.1 跳虫的采集与实验室饲养
供试跳虫分别采自生境不同的地点(表1),同时采集原栖息地表层土壤于饲养盒中,使用Baker氏酵母在实验室正常环境下饲养跳虫,并选择虫体基于形态学特征鉴定种名。
1.2 跳虫的饥饿处理与表面杀菌
从3种跳虫的饲养盒里分别取出20头成虫虫体于无菌培养皿中,置于室温环境下48 h左右,以保证跳虫体内的肠道菌群达到相对稳定的水平。将饥饿处理后的跳虫转移到4℃冰箱中,低温使其处于昏迷状态。在生物安全柜中用无菌水浸湿的小毛笔从培养皿中取出1头跳虫放在干净的载玻片上,将其首尾部位固定后,用小毛笔蘸取无菌水轻轻擦拭跳虫体表,以去除土粒和植物碎屑等。接着用75%的乙醇轻轻擦拭跳虫体表30 s,重复操作3~4次后,用无菌干滤纸吸去体表残留的乙醇。最后再用无菌水轻轻擦拭跳虫体表进一步去除残留的乙醇。

表1 3种跳虫的采集信息Table 1 Collecting data of three springtail species
1.3 跳虫的解剖与肠道内容物的收集
跳虫经过饥饿处理和表面杀菌后,使用镊子和微针刀在基恩士数码显微系统下对其胸、腹部进行解剖,并将跳虫肠道转移到无菌的干滤纸条上。同一滤纸条上连续收集20头同种跳虫的肠道及内容物后,将滤纸条转移到无菌离心管中,置于-40℃下保存、待用。
1.4 跳虫肠道菌群的16S rDNA扩增子测序
采用CTAB法提取3种跳虫肠道菌群的基因组DNA(Moranetal., 1993; 徐丽华等, 2007),测定DNA浓度并用琼脂糖凝胶电泳检测DNA的完整性。取适量的样本DNA于离心管中,使用无菌水稀释至1 ng/μL。以稀释后的基因组DNA为模板,针对16S rDNA V3-V4区,用带Barcode的特异性引物(341F: 5′-CCTAYGGGRBGCASCAG; 806R: 5′-GGACTACNNGGGTATCTAAT-3′),Phusion® High-Fidelity PCR Master Mix with GC Buffer和高效高保真酶(New England Biolabs公司)进行PCR。反应体系(50 μL): 2×Taq PCR Mix 25 μL, 引物341F/806R(10 μmol/L)各1 μL, DNA模板 2 μL, ddH2O 21 μL。反应条件: 95℃预变性5 min; 94℃变性1 min, 57℃退火45 s, 72℃延伸1 min, 共34个循环;最终72℃延伸10 min。PCR完成后,扩增产物使用2%的琼脂糖凝胶进行电泳检测,使用GeneJET 胶回收试剂盒(Thermo Scientific公司)剪切回收目标条带。PCR产物由北京诺禾致源科技股份有限公司在Ion S5TMXL平台上进行高通量测序。
1.5 生物信息学分析
IonS5TMXL下机数据首先使用Cutadapt对reads进行低质量部分过滤,再根据Barcode序列区分各样本数据,截去Barcode和引物序列初步质控后获得原始数据(raw reads)。接着通过与物种注释数据库比对并去除嵌合体序列,从而获得有效数据(clean reads)(Martin, 2011)。 利用Uparse软件对有效数据按97%的序列一致性进行操作分类单元(operational taxonomic units, OTUs)聚类分析(Rognesetal., 2016)。使用Mothur方法与SILVA132的SSU rRNA数据库对OTU序列进行物种注释(设定阈值为0.8~1),并分别在各分类层级上进行物种类群统计(Haasetal., 2011)。使用MUSCLE(v 3.8.31)软件进行快速多序列比对,获得所有OTU序列的系统发生关系(Wangetal., 2007)。基于均一化的OTU丰度信息,使用QIIME(v 1.9.1)和R软件(v 2.15.3)进行α和β多样性分析。将OTU代表序列通过Tax4Fun包比对到KEGG数据库(Asshaueretal., 2015),进而实现对菌群基因功能的预测。
2 结果
2.1 菌群测序质量与OTUs分布
3种跳虫肠道菌群测序的原始数据(raw reads)经进一步预处理后,从而获得最终的有效数据(clean reads),具体的数据产出统计及质控信息见表2。我们发现有效数据中碱基质量值大于20(测序错误率小于1%)的碱基比例均大于80%,且有效数据比例都超过85%,说明测序数据质量满足后续分析的要求。为了检验测序量是否足够,我们对测序数据进行了抽平处理,绘制出样本稀释曲线(图1: A)。结果发现随着测序深度的增加样本稀释曲线逐渐趋向于平坦,说明测序数据量基本覆盖到样本中的所有物种,更多的测序数据量对发现新OTU作用很小。从S. (C.)oligoseta,P.minuta和T.missus3种跳虫肠道菌群测序数据中我们分别获得了445, 621和632个OTUs。为了比较彼此之间OTUs的差异,利用R软件绘制出Venn图。由图1(B)可知,3种跳虫中共有的OTUs为141个,分别占S. (C.)oligoseta总OTUs的31.69%,占P.minuta总OTUs的22.71%,占T.missus总OTUs的22.31%。
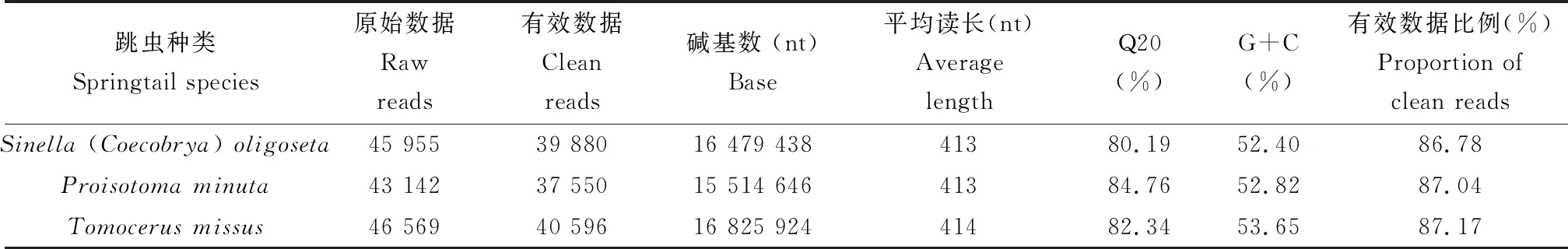
表2 3种跳虫肠道菌群测序数据预处理统计及质控信息Table 2 Statistics and quality control information of sequencing data preprocessedfor gut microbiota of three springtail species
Q20: 碱基质量分值大于20的reads的比例The proportion of reads with a base quality score greater than twenty.

图1 3种跳虫成虫肠道菌群OTUs稀释曲线(A)及Venn图(B)Fig. 1 Rarefaction curves (A) and Venn diagram (B) of the adult gut microbial OTUs in three springtail species
2.2 菌群α和β多样性分析
3种跳虫肠道菌群α多样性指数的统计(默认97%阈值)见表3。S. (C.)oligoseta肠道菌群的测定物种指数(445), Chao1指数(457.000)和ACE指数(456.495)最小,表明它的菌群丰富度最低。T.missus肠道菌群的香农指数(8.228)最大且辛普森指数(0.925)最小,表明它的菌群多样性最高。另外,S. (C.)oligoseta肠道菌群的谱系多样性指数(86.021)高于其他两种跳虫的相应指数,表明这种跳虫的肠道菌群对进化历史保存的差异最大,即菌群间亲缘关系最复杂,进化距离最远。
基于衡量β多样性指数的unifrac距离,我们检测了3种跳虫肠道菌群两两之间的相异系数(图2),结果发现T.missus和S. (C.)oligoseta肠道菌群存在的差异性较大,它们之间的weighted unifrac和unweighted unifrac距离分别为0.519和0.781。对3种跳虫肠道菌群的主成分分析(principal component analysis, PCA)和主坐标分析(principal co-ordinates analysis, PCoA)(图3: A, B),结果发现彼此之间的距离都比较远,表明它们的肠道菌群差异较大。为了更好地反映生态学数据的非线性结构,我们基于Bray-Curtis距离对测序数据执行无度量多维标定(non-metric multi-dimensional scaling, NMDS)分析。如图3(C)所示,Stress值小于0.001说明本次分析能准确反映3种跳虫肠道菌群的特点,且彼此之间的距离也比较远,这再次表明它们的肠道菌群存在较大的差异性。
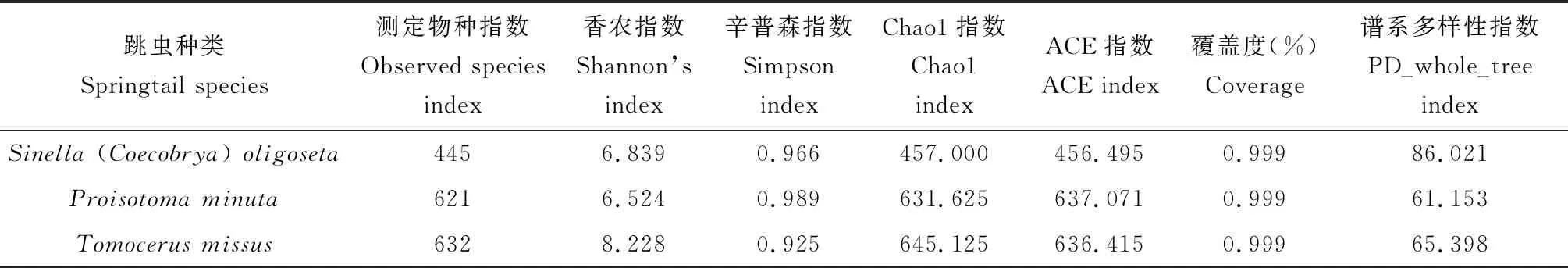
表3 3种跳虫肠道菌群α多样性指数Table 3 Alpha diversity indices of gut microbiota of three springtail species
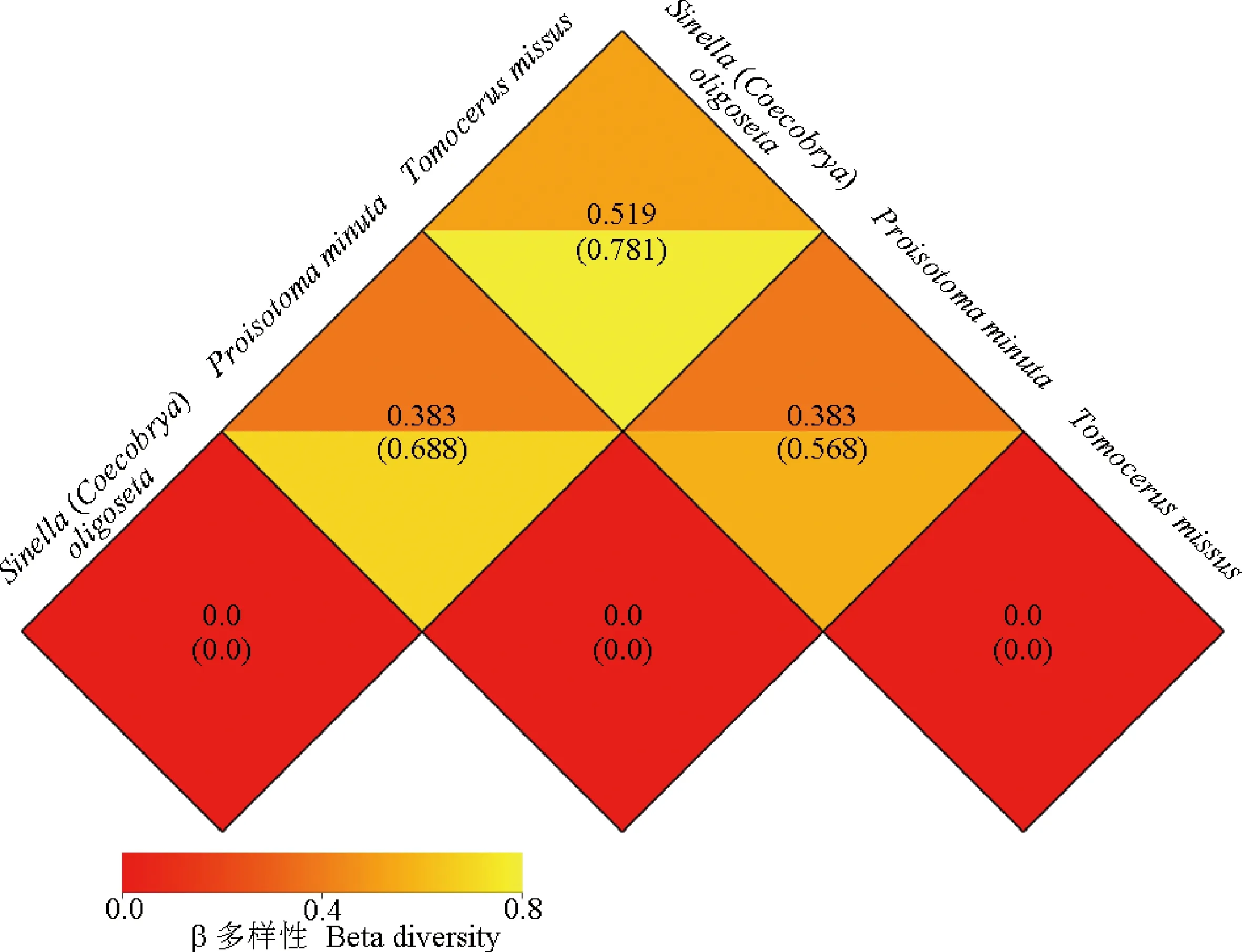
图2 3种跳虫肠道菌群的β多样性指数热图Fig. 2 Heatmap of beta diversity indices of gut microbiota of three springtail species图中方格中的数字是两两样本之间的相异系数,同一方格中上下两个值分别代表weighted unifrac和unweighted unifrac距离。The number in the square is dissimilarity coefficient between two samples. The two values on the top to bottom in the same square are weighted unifrac and unweighted unifrac distances, respectively.

图3 3种跳虫肠道菌群的主成分分析(PCA)(A)、主坐标分析(PCoA)(B)和无度量多维标定(NMDS)分析(C)Fig. 3 Principal component analysis (PCA) (A), principal co-ordinates analysis (PCoA) (B) and non-metricmulti-dimensional scaling (NMDS) analysis (C) for gut microbiota of three springtail speciesPC1: 第1主成分Principal component 1; PC2: 第2主成分Principal component 2; PCo1: 第1主坐标Principal coordinate 1; PCo2: 第2主坐标Principal coordinate 2; MDS1: 第1排序轴Dimensional scaling axis 1; MDS2: 第2排序轴Dimensional scaling axis 2.
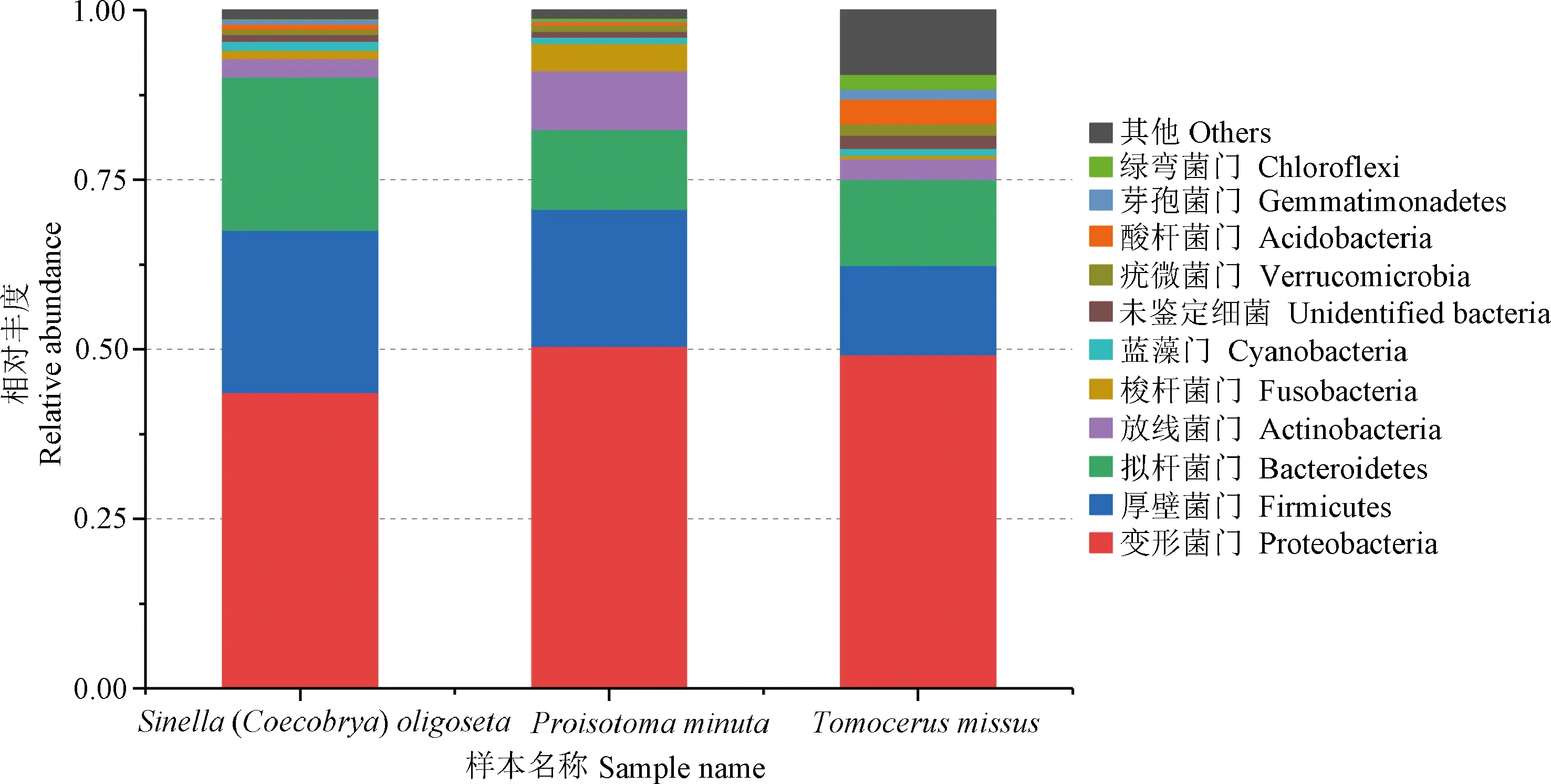
图4 3种跳虫肠道菌群在门水平上相对丰度前10的微生物类群Fig. 4 Relative abundance of top ten predominant microbiota at the phylum level in gut microbiota of three springtail species
2.3 菌群OTUs物种注释及相对丰度
对3种跳虫肠道菌群测序的OTUs进行物种注释(图4)。在门水平上S. (C.)oligoseta的肠道菌群主要属于变形菌门(Proteobacteria, 43.63%),厚壁菌门(Firmicutes,23.93%),拟杆菌门(Bacteroidetes, 22.63%),放线菌门(Actinobacteria, 2.64%),梭杆菌门(Fusobacteria, 1.33%),蓝藻门(Cyanobacteria, 1.3%),酸杆菌门(Acidobacteria, 0.8%),疣微菌门(Verrucomicrobia, 0.8%),芽孢菌门(Gemmatimonadetes, 0.58%),绿弯菌门(Chloroflexi, 0.08%)等19个门;P.minuta的肠道菌群主要属于变形菌门(Proteobacteria, 50.5%),厚壁菌门(Firmicutes, 20.21%),拟杆菌门(Bacteroidetes, 11.75%),放线菌门(Actinobacteria, 8.7%),梭杆菌门(Fusobacteria, 4.01%),疣微菌门(Verrucomicrobia, 0.96%),蓝藻门(Cyanobacteria, 0.95%),酸杆菌门(Acidobacteria, 0.48%),绿弯菌门(Chloroflexi, 0.34%),芽孢菌门(Gemmatimonadetes,0.11%)等21个门;T.missus的肠道菌群主要属于变形菌门(Proteobacteria, 49.23%),拟杆菌门(Bacteroidetes, 13.19%),厚壁菌门(Firmicutes, 12.65%),放线菌门(Actinobacteria, 3.05%),酸杆菌门(Acidobacteria, 3.6%),疣微菌门(Verrucomicrobia, 1.72%),绿弯菌门(Chloroflexi, 2.19%),芽孢菌门(Gemmatimonadetes, 1.52%),蓝藻门(Cyanobacteria, 0.96%),梭杆菌门(Fusobacteria, 0.55%)等29个门。根据以上数据,我们发现3种跳虫肠道菌群中变形菌门为最主要的类群,其次为厚壁菌门、拟杆菌门。另外,3种跳虫肠道菌群中都明显存在一定数量的放线菌。

图5 3种跳虫肠道菌群在属水平上相对丰度前30的微生物类群Fig. 5 Relative abundance of top thirty predominant microbiota at the genus level in gut microbiota of three springtail species
为了进一步比较3种跳虫肠道内优势菌属的差异,选取属水平丰度排名前30的类群生成堆积柱状图(图5)。我们发现它们的优势菌属包括假单胞菌属Pseudomonas, 节杆菌属Arthrobacter, 拟杆菌属Bacteroides, 寡养单胞菌属Stenotrophomonas, 弧菌属Vibrio, 双歧杆菌属Bifidobacterium, 多核菌属Polynucleobacter, 未鉴定毛螺菌科(unidentified Lachnospiraceae), 鲸杆菌属Cetobacterium, 不动杆菌属Acinetobacter, 芽孢杆菌属Bacillus, 鞘脂单胞菌属Sphingomonas, 苍白杆菌属Ochrobactrum, 微杆菌属Microbacterium, 纤维菌属Cellulosimicrobium等。其中,在S. (C.)oligoseta肠道中假单胞菌属Pseudomonas的丰度(16.21%)明显高于在P.minuta和T.missus肠道中的丰度(分别为0.87%和1.37%);在P.minuta肠道中弧菌属Vibrio的丰度(25.81%)明显高于在S. (C.)oligoseta和T.missus肠道中的丰度(分别为3.35%和0.004%)。值得注意的是在3种跳虫肠道中都检测到了一定丰度的气单胞菌属Aeromonas。
2.4 优势菌属物种丰度聚类分析
根据相对丰度前35菌属的物种注释及丰度信息绘制出聚类热图(图6),3种跳虫肠道中相对丰度前35的菌属在进化系统发生上聚集为3大类。在水平方向上每种菌属在3种跳虫体内的丰度明显不同,整体上可以发现3种跳虫肠道菌群的优势菌属是不同的。
2.5 菌群基因功能预测
对3种跳虫肠道菌群测序数据用Tax4Fun法进行菌群基因功能预测,实现了SILVA数据库注释的OTU物种分类与KEGG数据库中原核生物代谢功能的有效链接。在Level 1水平上,3种跳虫肠道菌群中KEGG pathway注释到代谢相关的基因丰度分别为44.34%, 44.89%和45.98%;与人类疾病相关的基因丰度分别为6.42%, 6.60%和6.76%。在Level 2水平上,3种跳虫肠道菌群基因与11类代谢过程相关(图7: A),其中涉及碳水化合物代谢的基因丰度最高,其次是涉及氨基酸代谢的基因。同时还发现菌群基因与8类疾病相关,其中涉及传染性疾病和耐药性的基因丰度明显高于其他功能基因的丰度(图7: B)。在Level 3水平上,3种跳虫肠道菌群中与代谢相关的丰度前20的基因其中涉及淀粉和蔗糖代谢、糖酵解/糖异生以及氨基糖和核苷酸糖代谢的基因在S. (C.)oligoseta肠道菌群中的丰度偏高。而涉及氨基酸相关酶等菌群基因在3种跳虫肠道内的丰度差别不明显(表4)。同时还关注了与人类疾病相关的菌群基因丰度前35的注释结果(表5),其中与β内酰胺抗药性、抗菌素耐药性和金黄色葡萄球菌感染相关的基因丰度在S. (C.)oligoseta肠道菌群中相对偏高;与胰岛素抗药性、幽门螺杆菌感染的上皮细胞信号传导和I型糖尿病相关的基因丰度在P.minuta肠道菌群中相对偏高;与军团病、亨廷顿氏病和非酒精性脂肪性肝病相关的基因丰度在T.missus肠道菌群中相对偏高。

图6 3种跳虫肠道菌群在属水平上相对丰度前35类群的聚类热图Fig. 6 Heatmap of top thirty-five predominant microbiota at the genus level in gut microbiota of three springtail species
3 讨论
本研究采用16S rDNA扩增子测序法对我国3个地区的3种跳虫的成虫肠道菌群进行了物种组成分析,结果发现S. (C.)oligoseta,P.minuta和T.missus肠道中的核心菌门均为变形菌门(Proteobacteria), 厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)。所以,我们认为在门水平上3种跳虫肠道菌群存在相似性。实际上早期研究也曾报道在白符跳F.candida和P.fimiata肠道(或粪便)中发现了一些变形杆菌、厚壁菌(Thimmetal., 1998; Hoffmannetal., 1999)。另外,在3种跳虫肠道中我们均检测出了一定比例的放线菌,其中包括微杆菌属Microbacterium, 双歧杆菌属Bifidobacterium, 纤维菌属Cellulosimicrobium等。近期Agamennone等(2018)用M490培养基从5种跳虫肠道内分离出链霉菌StreptomycesSc8, 纤维菌CellulosimicrobiumTm1, 微杆菌MicrobacteriumFc16a, 谷氨酸杆菌GlutamicibacterSc3等18种放线菌,并发现它们对细菌、真菌和卵菌病原体有一定的抗性。跳虫主要栖息于表层土壤、落叶层、苔藓等微生物为主的环境中(陈建秀等, 2007),它们对病原微生物的耐受性可能是体内的共生放线菌通过资源竞争或产生针对外源微生物的抗菌剂进行有效防御的结果。此外,王立秀等(2018)采用羧甲基纤维素钠选择性培养基从阿南原等跳肠道中筛选出阿里莱坦西节杆菌Arthrobacterarilaitensis, 烟草谷氨酸杆菌Glutamicibacternicotianae, 玉米白杆菌Leucobacterzeae3种放线菌,并发现它们有降解纤维素的能力。从跳虫肠道内分离放线菌不仅有益于新型放线菌的开发及其代谢产物的有效利用,而且有助于深入了解宿主抵御外界干扰适应复杂环境的能力。
通过本研究我们发现,S. (C.)oligoseta,P.minuta和T.missus3种跳虫肠道菌群存在较大的差异,其中T.missus肠道菌群的多样性相对最高,我们推测可能是因为该种跳虫栖息于生长茂盛的植被根部表层土壤,其中的微生物种类丰富,在此环境中长期的摄食活动增加了它的肠道菌群多样性。过去的研究已证明跳虫肠道是一个微生物选择性定植场所,摄食及食物偏好性会改变其肠道菌群结构(Hoffmannetal., 1999)。另外,Staubach等(2013)通过研究实验室和野生环境中果蝇的细菌菌群也表明环境因素(尤其食物来源)能最大程度影响宿主相关菌群结构。然而,由于我们选用的3种跳虫分别属于3个不同的科,其遗传背景差别较大,所以物种本身生理特点也会对肠道内微生物的定植产生一定的影响。实际上这3种跳虫的菌群结构与近期Agamennoe等(2019)报道的白符跳F.candida菌群结构确实存在较大差异。本研究3种跳虫肠道菌群的差异性还表现为在S. (C.)oligoseta肠道中假单胞菌属Pseudomonas的丰度(16.21%)明显高于在P.minuta和T.missus肠道中的丰度(分别为0.87%和1.37%),我们认为可能是因为S. (C.)oligoseta长期取食于阴暗潮湿的枯枝落叶层下,其中假单胞菌属的含量较高,而且有文献指出假单胞菌属与有氧环境中腐败动植物的降解有关(陈绍兴, 2005)。在P.minuta肠道中弧菌属Vibrio的丰度(25.81%)明显高于在S. (C.)oligoseta和T.missus肠道中的丰度(分别为3.35%和0.004%),其原因可能是它们采集于盐碱地水域附近的表层土壤,某些弧菌(如创伤弧菌)在这种环境下会发生富集作用(殷红秋等, 2016),而生活在该地区的跳虫对弧菌的摄食偏好性导致了其丰度的增加。总之,3种跳虫的肠道菌群结构存在差异性,不同栖息地环境与物种遗传背景是影响其菌群多样性的重要因素。

图7 Level 2水平上3种跳虫肠道菌群基因与代谢(A)及人类疾病(B)相关的KEGG pathway注释Fig. 7 Annotations of KEGG pathway related to metabolism (A) and human diseases (B)for the gut microbial genes in three springtail species at level 2
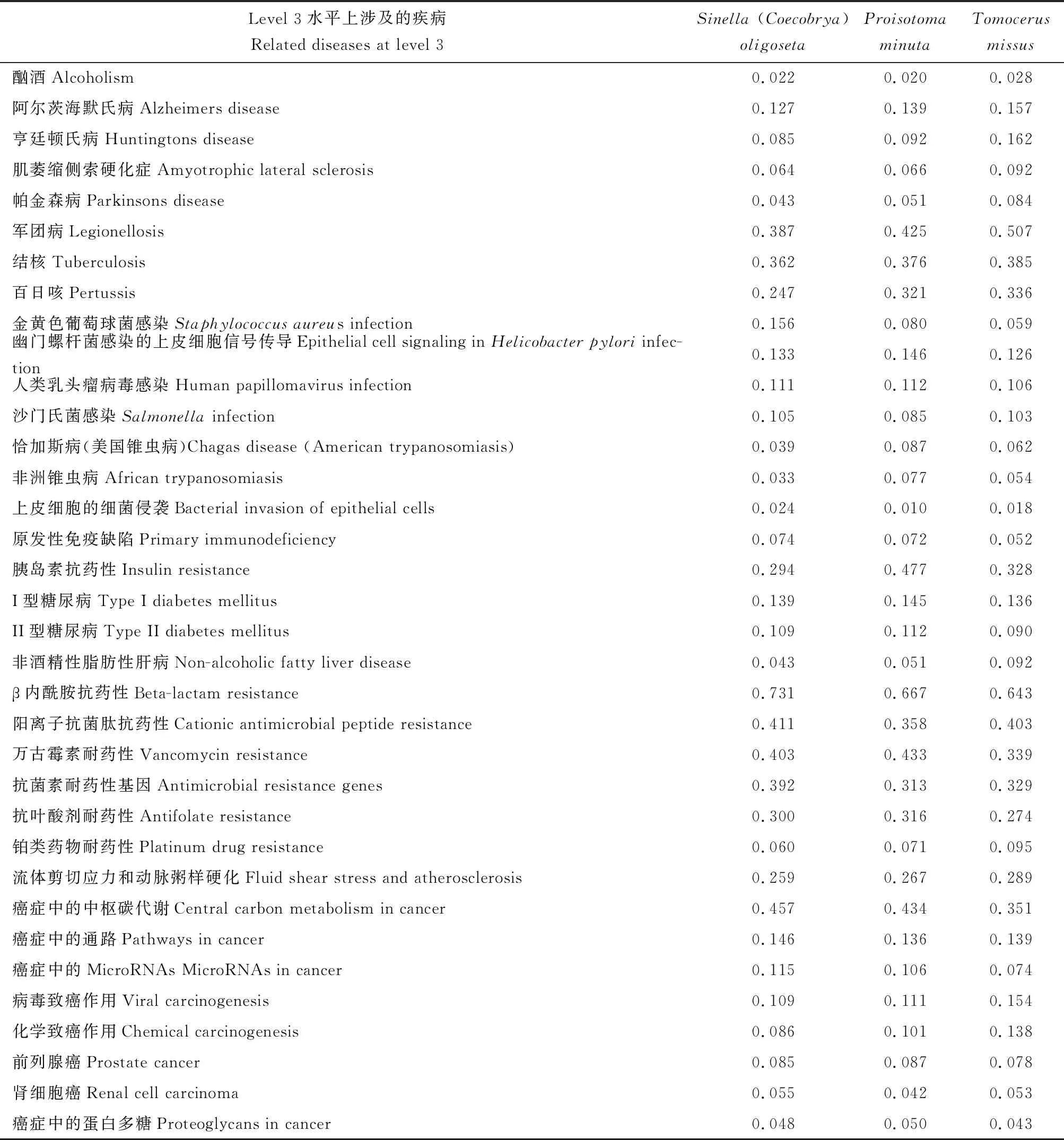
表5 KEGG pathway注释的Level 3水平上与人类疾病相关的3种跳虫肠道菌群基因的相对丰度(%)Table 5 Relative abundance (%) of gut microbial genes involved in human diseasesin three springtail species at level 3 in KEGG pathway annotation
通过Tax4Fun对3种跳虫肠道菌群基因的KEGG pathway注释,我们发现它们的菌群基因功能与碳水化合物代谢和氨基酸代谢明显相关,这与跳虫主要摄食和消化土壤中的动植物残骸和腐殖质等物质相对应(陈建秀等, 2007)。而且在长期生存于枯枝落叶层下的S. (C.)oligoseta肠道中涉及糖类物质代谢的菌群基因相对偏高,可能就是因为其肠道中存在大量与动植物降解有关的假单胞菌。另外,3种跳虫肠道菌群基因功能与人类疾病相关,其中涉及传染性疾病和耐药性的菌群基因丰度明显较高,这表明3种跳虫肠道内很可能存在耐药性和致病性(或潜在致病性)菌株。事实上我们的测序结果也检测出了气单胞菌属Aeromonas、苍白杆菌属Ochrobactrum和弧菌属Vibrio,相关研究已经报道这些菌属中存在耐药性或致病性菌株,如嗜水气单胞菌Aeromonashydrophila、人苍白杆菌Ochrobactrumanthropi、创伤弧菌Vibriovulnificus等(李皇, 2008; 刘志国等, 2015; 殷红秋等, 2016)。然而,为了进一步确定它们的肠道内是否存在耐药性和致病性细菌,还需要选择合适的培养基和培养条件进行菌株的分离和鉴定。此外,结果还预测出3种跳虫肠道菌群基因功能涉及内分泌与代谢疾病、神经退行性疾病和癌症等人类疾病,我们认为基于扩增子测序数据对菌群功能预测而获得的这一发现的可信度较低。因为这种菌群功能预测方法本身存在一定的局限性:首先是只能对已知微生物的已知功能进行预测;其次是预测结果受参考数据库的影响;另外原始数据预处理造成的部分数据损失也会影响预测结果(孙善峰等, 2019)。所以,在实际应用中肠道菌群基因的Tax4Fun功能预测不能取代全基因组来研究菌群功能,但是对于后续的实验设计和研究方向还是有着一定的指导意义。
总之,本研究表明S. (C.)oligoseta,P.minuta和T.missus3种跳虫肠道内的核心菌群为变形菌门、厚壁菌门和拟杆菌门,同时也存在较高丰度的放线菌。它们的差异性主要表现为菌群多样性和优势菌属相对丰度的不同,其影响因素包括物种自身遗传背景的差异,以及各自栖息地环境中微生物种类和数量的不同。
