甘薯G病毒吉林分离物的全基因组序列测定与分析
2020-06-08马俊丰李小宇张春雨周雪平王永志
马俊丰 李小宇 张春雨 周雪平 王永志

摘要 利用RT-PCR結合RACE方法,从感染SPVG的甘薯叶片中获得甘薯G病毒吉林分离物Jilin-gzl的全基因组序列。测序获得的Jilin-gzl分离物(GenBank No.Mk392509)基因组为10 797 nt。该病毒基因组含有一个10 464 nt的开放阅读框,编码由3 488个氨基酸构成的多聚蛋白。在P1和P3基因中也发现了由移码翻译产生的PISPO蛋白和PIPO蛋白。比对分析显示,在开放阅读框水平上Jilin-gzl与6个不同分离物核苷酸序列一致性为79%~99%,氨基酸一致性为92%~99%,其中与SC11、IS103、HG167和Jesus_Maria分离物一致性最高,与WT325和AI一致性最低。系统发育分析显示,Jilin-gzl与HG167和WCFR11系统发育关系最近,与中国台湾地区分离物WT325系统发育关系较远。这是中国大陆地区关于SPVG分离物全基因序列的首次报道,同时也是首次在中国东北地区发现SPVG。该研究探明了Jilin-gzl分离物的基因结构,系统发育关系,丰富了SPVG基因组序列信息,为后续开展SPVG种群的遗传进化及功能研究奠定了基础。
关键词 甘薯G病毒(SPVG); 全基因组; 比对分析; 系统发育
中图分类号: S 432.41
文献标识码: A
DOI: 10.16688/j.zwbh.2019163
Complete genome sequencing and analysis of Sweet potato
virus G isolate from Jilin, China
MA Junfeng1,2, LI Xiaoyu2, ZHANG Chunyu2, ZHOU Xueping3, WANG Yongzhi2
(1. Jilin Agricultural University, Changchun 130118, China; 2. Jilin Academy of Agricultural
Sciences, Gongzhuling 136100, China; 3. Institute of Plant Protection, Chinese Academy of
Agricultural Sciences, Beijing 100193, China)
Abstract
The complete genome of Jilin-gzl was determined by using rapid-amplification of cDNA ends (RACE) and RT-PCR. The complete sequence of Jilin-gzl (GenBank No.Mk392509) had 10 797 nucleotides, excluding the 3′-terminal poly (A) tail. Its genome contained an open reading frame of 10 464 nucleotides and encoded a polyprotein of 3 488 amino acids. Two additional proteins, termed ‘PISPO and ‘PIPO, were also translated by frame shifting within the P1 and P3 cistron. The results of sequence identity analysis of Jilin-gzl isolate with the 6 reference SPVG isolates showed that Jilin-gzl isolate shared a 79%-99% nucleotide identity and 92%-99% amino acid identity with other SPVG isolates at the ORF sequence level. Jilin-gzl isolate shared a highest sequence identity with SC11, IS103, HG167, Jesus_Maria isolates, and a lowest identity with WT325 and AI isolates. Phylogenetic analysis indicated that Jilin-gzl shared high sequence homology with HG167 and WCFR11 isolates, and shared low sequence homology with WT325 isolate from Taiwan, China. This work was the first report about the complete sequence of SPVG in Mainland China and it was also the first time that SPVG has been found in Northeast China. This research elucidated the genetic structure and phylogenetic relationship of Jilin-gzl isolate, and enriched the information of SPVG genome sequence. These will provide useful information for further study of the phylogenesis and function of this pathogen.
Key words
Sweet potato virus G (SPVG); complete genome; comparative analysis; phylogeny
甘薯Ipomoea batatas为旋花科Convolvulaceae薯蓣属Dioscorea代表植物,是中国主要粮食作物之一。中国是世界第一大甘薯生产国[1],据联合国粮食及农业组织(FAO)统计,2017年中国甘薯总产量约7 203万t,占世界甘薯总产量(11 283万t)的63.8%。甘薯作为无性繁殖作物,主要通过块茎进行繁殖,这种繁殖方式极易积累病毒。被侵染后的甘薯,其产量和品质大幅降低,造成严重的经济损失。据Clark等2012年报道,在世界范围内已发现的能够侵染甘薯的病毒共有30多种[2],其中有20多种病毒已经在中国被发现[39]。甘薯G病毒Sweet potato virus G(SPVG),是马铃薯Y病毒科Potyviridae马铃薯Y病毒属Potyvirus成员之一,于1994年最早在中国被发现,之后美国、秘鲁、韩国、阿根廷等国家也相继报道了该病毒[1013]。SPVG在自然条件下通过蚜虫以非持久性方式传播,同时也可以通过汁液摩擦、嫁接等机械方式传播。近几年在中国各个甘薯种植区,SPVG时有发生,并且已成为影响甘薯品质与产量的重要病毒之一。
SPVG基因组由正单链RNA组成,含有一个较大开放阅读框(open reading frame,ORF),编码一个多聚蛋白(polyprotein)。基因组5′和3′端各有一段非编码区(untranslated regions,UTRs),3′非编码区末尾具有poly(A)尾序列。多聚蛋白经自身编码的蛋白酶切割后,得到不同的功能蛋白。除此之外,SPVG基因组中还有两个小的开放阅读框,通过移码翻译(read frame shift)的方式编码蛋白PISPO和蛋白PIPO,这两个蛋白分别与P1和P3蛋白的N端以P1N-PISPO和P3N-PIPO融合形式存在[14]。SPVG基因组通过这两种方式生成12个成熟的功能蛋白,使各个基因呈现不同程度的遗传多样性。目前,中国大陆地区关于SPVG基因组序列的报道仅集中在部分基因区段,如外壳蛋白(CP)基因等[15],尚未见有关SPVG全基因组序列的报道。本次研究拟通过对SPVG吉林分离物Jilin-gzl的全基因序列测定,对其序列特征、系统发育关系等进行系统分析,为后续开展SPVG相关功能研究奠定基础。
1 材料与方法
本试验于2018年11月至2019年1月在吉林省农业科学院植物保护研究所微生物实验室完成。
1.1 试验材料
SPVG吉林分离物Jilin-gzl于2018年9月采集自吉林省长春市甘薯种植区,呈典型明脉、花叶症状,前期经血清学检测和CP基因扩增初步确定为SPVG分离物,将新鲜样品置于密封袋中,于-80℃冻存。
1.2 主要试剂
RNeasy Plant Mini Kit为QIAGEN公司产品,ReverAid First Strand cDNA Synthesis Kit为赛默飞世尔科技公司(Thermo Fisher Scientific)产品,AxyPrep DNA Gel Extraction Kit为爱思进生物技术公司(Axygen)产品,SMARTer RACE 5′/3′ Kit、TaKaRa LA Taq、pMD18-T Vector Cloning Kit、RNA Marker、DNA Marker为宝生物工程公司(TaKaRa)产品。
1.3 试验方法
1.3.1 RNA提取及基因组克隆
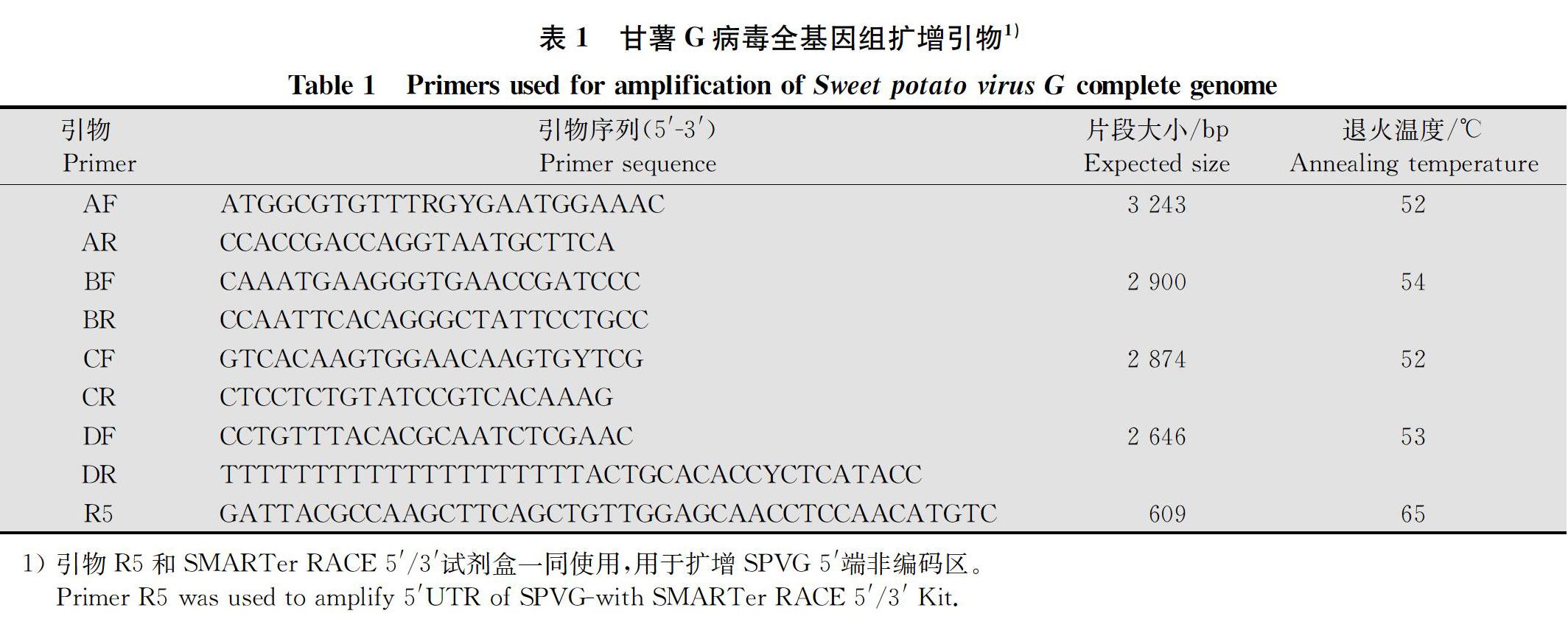
采用TRIzol试剂法从确定感染SPVG的甘薯叶片中提取总RNA,提取步骤参照RNeasy Plant Mini试剂盒(QIAGEN)说明书。以总RNA为模板,参照反转录试剂盒说明书进行反转录,反应体系:Oligo dT Primer 1 μL、dNTP Mixture 1 μL、Total RNA 1 μL、RNase Free H2O 7 μL,于65℃温浴5 min,然后立即冰上冷却,并依次加入:RNase Inhibitor (40 U/μL) 0.5 μL、PrimeScript Ⅱ RTase (200 U/μL) 1 μL、5×PrimeScript Ⅱ Buffer 4 μL,30℃反应10 min,42℃反应60 min。根据GenBank中已登录的12个SPVG不同分离物(登录号及名称见图3)全基因序列的保守区,设计用于合成全基因序列的简并引物和特异性引物(表1),以上引物委托吉林省库美生物科技有限公司合成。
PCR扩增采用50 μL反应体系:TaKaRa LATaq(5 U/μL) 0.5 μL、10×LA PCR Buffer Ⅱ (Mg2+Plus) 5 μL、dNTP Mixture 5 μL、cDNA 1 μL、正向引物(10 μmol/L)1 μL、反向引物(10 μmol/L)1 μL、超纯水36.5 μL。PCR反应体系:94℃预变性5 min;94℃变性30 s,退火30 s(退火溫度视基因片段而定,表1),72℃延伸若干分钟(延伸时间按1 kb/min计算),共30个循环;最后72℃再延伸5 min。
PCR反应结束后,取3 μL产物用1%琼脂糖凝胶电泳进行检测,检测后切取目的条带,利用胶回收试剂盒进行纯化,纯化产物与pMD18-T载体进行连接,并转化到E.coli DH5α感受态细胞中。通过菌落PCR筛选出阳性克隆子,随机选取3~5个,委托吉林省库美生物科技有限公司进行测序。根据测序结果设计用于扩增5′非编码区的引物(表1),5′非编码区的扩增采用SMARTer RACE 5′/3′试剂盒(TaKaRa),步骤依照说明书进行。
1.3.2 序列分析
利用DNAMAN软件进行序列拼接,在网站http:∥www.dpvweb.net/potycleavage/index.html上进行多聚蛋白剪切位点分析,在http:∥blast.ncbi.nlm.nih.gov/上進行序列一致性分析。
1.3.3 系统发育分析
为探究SPVG不同分离物的系统发育关系,从GenBank中选取11个其他SPVG分离物的核苷酸序列作为参考,使用最大似然法(maximum likehood, ML)基于全基因序列编码区(coding sequence, CDS)核苷酸序列构建SPVG系统发育树。建树之前,采用DAMBE软件检测核苷酸序列替换是否饱和。利用MEGA 6软件选择最优化的核苷酸替换模型,并参照BIC(Bayesian information criterion)标准设置相应参数,最后通过自举法(bootstrap)来对各分支节点的置信度(bootstrap confidence level)进行评估。
2 结果与分析
2.1 Jilin-gzl分离物的全基因组序列特征
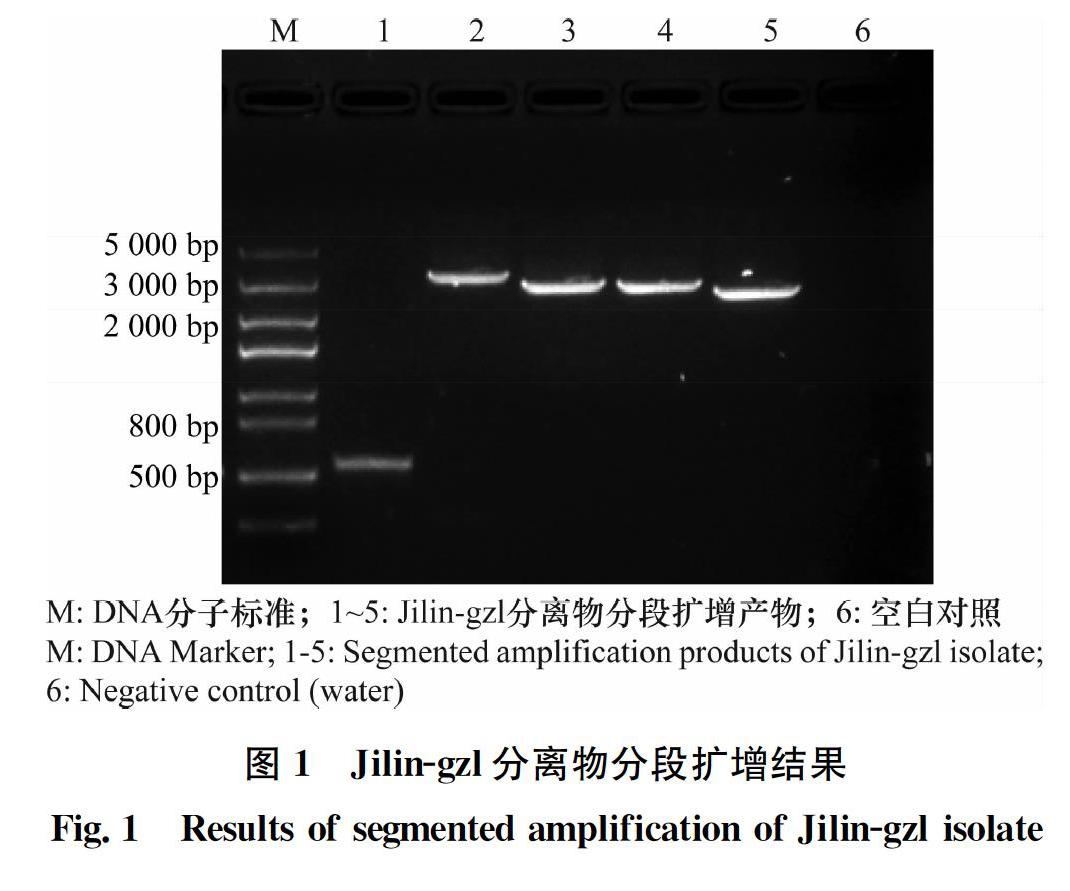
Jilin-gzl分离物全基因组分段扩增结果如图1所示,各片段大小均符合预期。
通过测序、拼接获得的Jilin-gzl分离物基因组全长为10 797 nt (GenBank No.Mk392509),5′端和3′端非编码区(untranslated regions, UTRs)的长度分别为113 nt和238 nt,其中3′末端具有poly(A)尾序列(图2)。该分离物仅包含一个开放阅读框,位于11410 577 nt,翻译出一个包含3 488个氨基酸残基的多聚蛋白,属于典型的Potyvirus属病毒的基因结构。剪切位点分析显示,Jilin-gzl分离物的9个剪切位点分别为PYMEQY/S、KHYLVG/G、DEVQHQ/A、GPVYHQ/S、NCVQHQ/S、EIVQHQ/A、TVVEHE/S、VPVYTQ/S、NNVHHQ/S。Jilin-gzl分离物不同基因的位置、大小以及编码蛋白的大小如表2所示。
2.2 Jilin-gzl分离物的序列一致性分析
序列一致性分析显示,在开放阅读框水平上Jilin-gzl与其他6个分离物的核苷酸和氨基酸相似性分别为79%~99%、92%~99%(表2),其中与Jesus_Maria、SC11、IS103、HG167相似性最高,均为99%,与WT325相似性最低,仅为79%和88%。在5′和3′非编码区上,核苷酸一致性分别为65%~98%和91%~99%,其中在5′非编码区上Jilin-gzl与Jesus_Maria、WT325一致性较低,仅为76%和65%,而与SC11、IS103、HG167、AI一致性较高,均超过90%;在3′非编码区上一致性较高,均超过90%。在单个基因区段上Jilin-gzl与Jesus_Maria、SC11、IS103、HG167的核苷酸和氨基酸一致性分别为98%~99%和97%~100%,一致性均非常高;而与WT325和AI的核苷酸和氨基酸一致性为66%~87%和37%~97%,一致性较低。综上所述,Jilin-gzl与分离物SC11、IS103、HG167、Jesus_Maria一致性非常高,与WT325、AI一致性较低。
2.3 系统发育分析
建树序列经过多重比对与Gblock剪裁后,得到长度为10 231 bp的序列保守区。根据MEGA软件的BIC标准,建树序列最合适的核苷酸替换模型为GTR+I+G。ML法构建的系统发育树如图3所示,12个分离物共形成4个区域,其中系统发育关系较近的分离物聚为一簇,GWB-2(JN613807,SPVG2)和SPFMV-UNB-01(MF185715,SPFMV)为外族。从图中可以看到,区域Ⅲ的各个分离物并未像区域Ⅰ和区域Ⅱ那样聚为一簇,但是从图中的拓扑结构来看,Jilin-gzl与HG167和WCFR11系统发育关系最近,而与该区域其他分离物系统发育关系相对较远。同时Jilin-gzl与中国台湾地区分离物WT325系统发育关系相对较远而与两个国外分离物系统发育关系较近,表明SPVG各分离物在遗传多样性上与地理分布无显著联系。
3 结论与讨论
本研究通过克隆、测序等手段获得了SPVG吉林分离物Jilin-gzl的全基因组序列,并且通过序列一致性比对,构建系统发育树等方法对其基因组结构特征及其系统发育关系进行了全面的探究与分析。这是中国大陆地区首次关于SPVG全基因组序列的报道,同时也是首次在中国东北地区发现SPVG。本研究结果为后续深入开展SPVG种群的遗传进化及相关功能研究奠定了基础。
序列一致性比对分析结果中有一部分数据值得注意,首先在Jilin-gzl与Jesus_Maria和AI比对结果中发现,Jilin-gzl与Jesus_Maria在5′非编码区核苷酸序列差异性较大,而在编码区和3′非编码区核苷酸序列差异性较小,与此相反的是,Jilin-gzl与AI在5′和3′非编码区核苷酸序列差异性较小,在编码区核苷酸序列差异性较大,类似数据差异在同为Potyvirus的马铃薯Y病毒Potato virus Y(PVY)全基因组序列一致性比对分析中也有发现,并且研究已表明这种数据差异是由PVY不同株系重组造成的[16],据此推断,Jilin-gzl分离物的形成也可能与不同SPVG分离物的重组有关。其次,在Jilin-gzl与WT325和AI的比较中发现,Jilin-gzl与这两个分离物在全基因组水平上序列差异较大,同时在系统发育树中也发现WT325和AI这两个分离物与Jilin-gzl系统发育关系较远,因此,SPVG可能存在多种株系。除此之外,本试验基于SPVG全基因组序列构建系统发育树,相较单基因序列建树,全基因组序列建树能够从基因组水平上,更准确地探究不同分离物之间的系统发育关系。
参考文献
[1] 马代夫, 李强, 曹清河, 等. 中国甘薯产业及产业技术的发展与展望[J]. 江苏农业学报, 2012, 28(5): 969973.
[2] CLARK C A, DAVIS J A, ABAD J A, et al. Sweet potato viruses: 15 years of progress on understanding and managing complex diseases [J]. Plant Disease, 2012, 96(2): 168185.
[3] 黃艳岚, 张超凡, 邹学校. 甘薯杆状病毒湖南分离物基因组全序列测定及比较分析[J]. 植物病理学报,2019,49(6):790798.
[4] 黄艳岚, 张道微, 董芳, 等. Small RNA深度测序鉴定甘薯种质的甘薯曲叶病毒[J]. 分子育种,2019, 17(11): 36413649.
[5] 刘起丽, 张建新, 李学成, 等. 侵染甘薯的DNA病毒研究进展[J]. 植物保护, 2017, 43(3): 3642.
[6] 范平民. 广西甘薯病毒病的检测及2种病毒(SPFMV和SPLCV)的基因组序列分析[D]. 南宁: 广西大学, 2016.
[7] 秦艳红, 渠瑞娜, 乔奇, 等. 甘薯病毒C中国分离物全基因组序列测定及比较分析[J]. 植物病理学报, 2018, 48(3): 324329.
[8] 乔奇, 张振臣, 张德胜, 等. 中国甘薯病毒种类的血清学和分子检测[J]. 植物病理学报, 2012, 42(1): 1016.
[9] 王爽, 田雨婷, 乔奇, 等. 侵染甘薯的菜豆金色花叶病毒属病毒和甘薯褪绿矮化病毒多重PCR检测方法的建立与应用[J]. 植物保护学报, 2018, 45(6): 14271428.
[10]SOUTO E R, SIM J, CHEN J, et al. Properties of strains of Sweet potato feathery mottle virus and two newly recognized potyviruses infecting sweet potato in the United States[J]. Plant Disease, 2007, 87(10):12261232.
[11]UNTIVEROSM, FUENTES S, KREUZE J. Molecular variability of sweet potato feathery mottle virus and other potyviruses infecting sweet potato in Peru [J]. Archives of Virology, 2008, 153(3):473483.
[12]KWAK H R, KIM J, KIM M K, et al. Molecular characterization of five potyviruses infecting Korean sweet potatoes based on analyses of complete genome sequences [J]. Plant Pathology Journal, 2015, 31(4):388401.
[13]RODRGUEZ PARDINA P E, BEJERMAN N, LUQUE A V, et al. Complete nucleotide sequence of an Argentinean isolate of Sweet potato virus G [J]. Virus Genes, 2012, 45(3): 593595.
[14]OLSPERT A, CARR J P, FIRTH A E. Mutational analysis of Potyviridae, transcriptional slippage site utilized for expression of the P3N-PIPO and P1N-PISPO proteins [J]. Nucleic Acids Research, 2016, 44(16): 76187629.
[15]古英洪, 汤浩茹, 张义正. 甘薯G病毒外壳蛋白基因克隆与序列分析[J]. 中国农学通报, 2006(9): 5055.
[16]CHANG Fei, GAO Fangluan, SHEN Jianguo, et al. Complete genome analysis of a PVYN-Wi recombinant isolate from Solanum tuberosum in China [J]. Potato Research, 2015, 58(4): 377389.
(责任编辑:田 喆)
收稿日期: 20190402 修订日期: 20190601
基金项目:国家重点研发计划(2017YFD0201604);吉林省农业科技创新工程(CXGC2017JQ021);吉林省农业产业技术体系马铃薯(甘薯)综合栽培技术示范与推广(2018)
致 谢: 参加本试验部分工作的还有江代礼、谭翰杰、张能和纪烨斌等同学,特此一并致谢。
通信作者 E-mail:周雪平zzhou@zju.edu.cn; 王永志yzwang@126.com
# 为并列第一作者
