过渡金属催化sp3C-H键活化构筑sp3C-P键反应研究进展
2020-06-05全丽霞何洪红祝显虹计从斌周安西戚丽毛刘量
全丽霞,何洪红,祝显虹,计从斌,周安西,戚丽,毛刘量
(上饶师范学院 化学与环境科学学院,江西 上饶 334001)
过去几十年,过渡金属催化的C-H键活化反应由于具备高化学区域选择性、高原子经济性和环境友好性,而备受化学研究工作者的青睐,并取得了显著的成效,已然成为高化学区域选择性构建碳-碳和碳-杂原子键的重要合成方法[1]。其中,由于sp3C-H的高度惰性,导致过渡金属催化sp3C-H键活化构筑碳-碳和碳-杂原子键,尤其是sp3C-P键的构筑,一直是过渡金属催化sp3C-H键活化研究领域的难点和热点。
有机磷(尤其是含sp3碳-磷键)化合物是一类非常重要的有机化合物,不仅广泛应用于天然产物、药物活性中间体和功能材料分子领域,而且在有机合成化学中,往往扮演着至关重要的角色,作为配体调控中心金属电性或催化剂促进有机反应的进行(图1)[2]。因此,sp3碳-磷键构筑反应的研究,一直是化学工作者研究关注的热点和难点。构筑sp3碳-磷键的方法主要有:(1)磷氢试剂对不饱和键亲核加成反应[3];(2)自由基串联反应[4];(3)过渡金属催化sp2/sp碳-磷键氢化反应[5],但这些反应并不是真正意义上的sp3C-H键活化构筑sp3C-P键,并且往往伴随着诸如化学区域选择性差、官能团兼容性较差、 底物结构局限性大等

图1 含sp3碳-磷键结构单元的生物活性分子和配体
缺点。基于对现代有机合成化学绿色高效发展理念的考虑,寻找一类简单、高效的催化体系来活化sp3C-H键构筑相应的碳-磷键显得尤为迫切和具有重要的研究价值。近些年来,通过过渡金属催化sp3C-H键活化构筑C-P键的反应研究也取得了重要进展。根据不同类型过渡金属催化剂进行分类,本文简要综述了近年来过渡金属催化sp3C-H键活化构筑sp3C-P键反应的研究进展,并对该研究领域的局限性和未来发展前景进行总结和展望。
1 铜催化sp3C-H键活化构筑sp3C-P键反应
2009年,李朝军小组报道以溴化亚铜为催化剂,氧气为氧化剂,实现了N-芳基四氢异喹啉sp3C-H键活化构筑C-P键的反应,高化学区域选择性合成了具有重要应用价值的α-氨基磷酸酯类化合物(图2)[6]。反应可能的历程为:首先氧气和铜盐协同催化氧化将N-芳基四氢异喹啉转化成亚胺正离子,随后弱亲核性五价磷异构成强亲核性的三价膦,并对亚胺正离子中间体进行亲核进攻,从而得到目标产物。该催化体系反应条件温和,以绿色环保、经济安全的氧气为氧化剂,官能团兼容性较好,但反应磷源只局限于亚磷酸酯,且四氢异喹啉氮上的取代基只能是芳基或取代芳基。

图2 铜催化N-芳基四氢异喹啉sp3C-H磷酰化反应
2015年,Miranda和García 报道了以溴化亚铜为催化剂,DDQ (2,3-二氯代-5,6-二氰基-1,4-苯醌)为氧化剂,乙腈和甲苯体积比为1∶1混合溶剂为反应溶剂,实现了N-甲基-5,6-二氢苯并菲啶和亚磷酸酯磷源的脱氢氧化偶联反应,高效合成具有抗菌活性的6-磷酰化-N-甲基-5,6-二氢苯并菲啶(图3)[7]。虽然该反应只局限于亚磷酸酯类磷源,但反应突破了胺类底物氮上取代基只能为吸电子基的局限,且反应同样经历亚胺正离子中间体的历程。值得注意的是,N-甲基-5,6-二氢苯并菲啶形成亚胺正离子中间体是具有化学区域选择性的,一种是形成环内亚胺正离子中间体,一种是形成环外亚胺正离子中间体。该小组通过调控反应条件可以化学区域选择性专一地形成环内亚胺正离子中间体,从而实现N-甲基-5,6-二氢苯并菲啶sp3C-H键活化构筑C-P键。

图3 铜催化N-甲基-5,6-二氢苯并菲啶sp3C-H磷酰化反应
同年,雷爱文小组报道了芳基肟醚与磷氧氢试剂的双自由基高效选择性偶联反应,成功构筑sp3C-P 键,合成具有潜在生物活性的β-酮磷酸酯类化合物(图4)[8]。该反应底物适用范围广、官能团兼容性高,并且反应能够同时兼容二芳基磷氧氢和亚磷酸酯两种磷源。值得注意的是,反应中添加剂无水乙酸酐对反应收率起着至关重要的作用,主要是为了稳定α-磷酰化亚胺中间体。
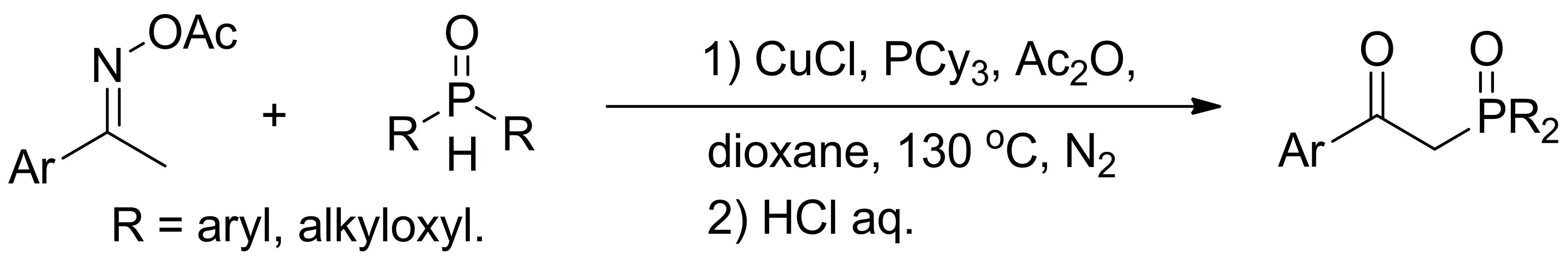
图4铜催化芳基肟醚sp3C-H磷酰化反应
该小组经过进一步研究,认为反应可能经历如下历程(图5):首先Cu(I)被芳基肟醚氧化成Cu(II),同时自身转化为亚胺氮自由基,而亚胺氮自由基快速异构成sp3C 自由基;随后磷氧氢试剂被Cu(II)氧化成磷自由基;接着sp3C自由基和磷自由基高效选择性交叉偶联得到α-磷酰化亚胺(可逆反应),接下来无水乙酸酐稳定α-磷酰化亚胺,使平衡向产物方向移动,生成烯胺中间体,最后经由水解得到产物β-酮磷酸酯化合物。

图5 铜催化芳基肟醚sp3C-H磷酰化反应机理
2016年,李朝军小组报道了以CuI为催化剂,TBHP(叔丁基过氧化氢)为氧化剂,实现了N-芳基甘氨酸酰胺衍生物和亚磷酸酯的脱氢氧化偶联反应,合成了α-/β-氨基磷酸酯类化合物(图6)[9]。该反应反应条件温和,室温下即可以良好的收率得到目标分子,但只有亚磷酸酯类磷源适用于此反应体系,且N-芳基甘氨酸酰胺底物中芳基上取代基只能是邻对位强富电子性的甲氧基。值得注意的是,甘氨酸酰胺衍生物底物中酰胺的氮上必须有活泼氢,反应才能顺利进行,主要可能是由于在反应中,酰胺的N-H参与过渡金属的配位作用,从而活化了甘氨酸酰胺衍生物底物而利于反应的进行。

图6 碘化亚铜催化甘氨酸酰胺衍生物sp3C-H磷酰化反应
该小组提出如下反应机理(图7):首先甘氨酸酰胺底物在催化剂和氧化剂的共作用下,形成亚胺金属配合物A,随后A经过分子内配体交换生成五元环铜螯合物B,接着磷亲核试剂对B进行亲核进攻,最后生成产物C,并释放出催化剂铜从而完成催化循环。

图7 碘化亚铜催化甘氨酸酰胺衍生物sp3C-H磷酰化反应机理
2 银催化sp3C-H键活化构筑sp3C-P键反应
2016年,高玉兴小组报道了N-H不保护四氢异喹啉类底物、醛和亚磷酸酯类磷源的三组分反应,一步构建sp3C-P键,高效合成磷酰化异喹啉类化合物(图8)[10]。该反应不但催化体系不需要氧化剂的参与,显得简洁高效,而且底物廉价易得、适用范围广、官能团兼容性好。另外,该反应具有优秀的化学区域选择性,只高选择性地得到异喹啉类底物C1位置磷酰化的产物。文章中该小组认为反应可能经历如下历程:首先,在醛的诱导作用和Ag(I)的协同催化下,底物四氢异喹啉脱去一分子水形成中间体环外亚胺金属正离子,而环外亚胺金属正离子会快速异构成热力学更稳定的环内亚胺金属正离子,随后弱亲核性五价磷异构成强亲核性的三价膦物种,并对环内亚胺金属正离子进行亲核进攻,生成目标产物并释放出催化剂Ag(I),从而完成催化循环。其中由环外亚胺金属正离子转化成热力学更稳定的环内亚胺金属正离子中间体是该反应具有专一化学区域选择性的关键。

图8 银催化四氢异喹啉sp3C-H磷酰化反应及其机理
2017年,董嘉兴小组报道了1,3-二羰基化合物和亚磷酸酯类磷源的氧化脱氢偶联反应,合成一系列官能团化的β-酮磷酸酯化合物(图9)[11]。该反应以醋酸银为催化剂,过二硫酸钾为氧化剂,在温和条件下实现1,3-二羰基化合物选择性sp3C-sp2C键裂解,高效构筑了sp3C-P键。反应底物适用范围广,底物简单易得,官能团兼容性强,且反应具有良好的区域选择性。反应中使用体积比(DMF∶H2O = 1∶1)混合溶剂作为反应溶剂,有助于氧化剂的溶解,从而提高反应收率。

图9 银催化1,3-二羰基化合物sp3C-H磷酰化反应
该小组在机理探究实验中,利用自由基捕捉剂TEMPO捕捉到了相应的自由基中间体。基于此文中提出该反应的历程(图10):首先,亚磷酸酯在醋酸银和过二硫酸钾的共作用下产生磷自由基,随后磷自由基对1,3-二羰基化合物的烯醇式进行加成生成三级碳自由基,与此同时,Ag(I)对三级碳自由基进行氧化并对其的双羰基进行配位,此时,体系中的水作为亲核试剂对银离子配合物进行亲核进攻,并同时伴随着一分子HOAc的离去,得到烯醇式产物,最后,烯醇式产物转化成最终产物β-酮磷酸酯化合物。
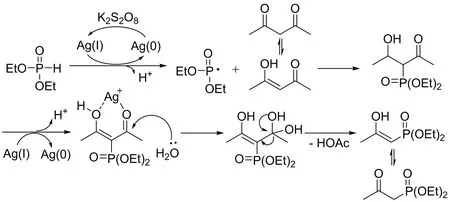
图10 银催化1,3-二羰基化合物sp3C-H磷酰化反应机理
3 铁催化sp3C-H键活化构筑sp3C-P键反应
2010年,Ofial小组报道了N,N-二烷基苯胺和亚磷酸酯类磷源的脱氢氧化偶联反应,以廉价的FeCl2为催化剂,过氧叔丁醇为氧化剂,在室温下以专一化学区域选择性构建sp3C-P键,合成一系列具有生物活性的α-氨基磷酸酯类化合物(图11)[12]。反应底物适用范围广,官能团兼容性好,反应磷源还能兼容环状亚磷酸酯类磷源。值得注意的是,该反应以甲醇为溶剂,利于稳定亚胺正离子中间体,从而提升反应产率。

图11 铁催化N,N-二烷基苯胺sp3C-H磷酰化反应
该小组在机理探究实验中,采用N-甲氧甲基-N,4-二甲基苯胺作为反应物,在模板反应条件下(剔除催化剂)直接和亚磷酸二乙酯反应,能以86%的产率得到目标产物。基于此,该小组提出一个可能的反应历程(图12):首先N,N-二烷基苯胺在催化剂和氧化剂的共同作用下生成亚胺正离子中间体A,中间体A在甲醇的溶剂化作用下,通过一个动态平衡转化成N-甲氧甲基-N-甲基芳胺B用以稳定中间体A,随后,五价磷源转化成亲核能力更强的三价磷源,并对亚胺中间体A进行亲核进攻,伴随着一个质子的离去,从而得到目标产物。

图12 铁催化N,N-二烷基苯胺sp3C-H磷酰化反应机理探究实验及其机理
4 钴催化sp3C-H键活化构筑sp3C-P键反应
2018年,唐果小组报道了三级胺和磷氧氢试剂的脱氢偶联反应,以廉价易得的醋酸钴和N-羟基邻苯二甲酰亚胺为共催化剂,空气为氧化剂,高效实现了三级胺sp3C-H的磷酰化反应,构建了sp3C-P键,合成一系列官能团化的α-氨基磷酸酯类化合物(图13)[13]。该反应底物适用范围广,三级胺类底物能够同时兼容链状烷基三级胺以及异喹啉类三级胺底物,且磷源能同时兼容亚磷酸和二芳基磷氧氢。值得注意的是,N-羟基邻苯二甲酰亚胺作为共催化剂,主要是为了改变钴催化剂中心金属的电性,从而提高反应的效率。

图13 钴催化三级胺sp3C-H磷酰化反应
在机理验证实验时,该小组发现在模板反应条件下,加入自由基捕捉剂TEMPO,反应并未被完全抑制。基于此,该小组提出一个可能的反应历程(图14):首先,醋酸钴和N-羟基邻苯二甲酰亚胺进行螯合形成中间体A,随后中间体A在氧气的氧化下生成三价钴中间体B,接着,三级胺作为路易斯碱和中间体B配位形成中间体C,随后中间体C进行分子内单电子转移形成中间体D,而后,中间体D中的氧自由基攫取三级胺α位上的氢,伴随催化剂的离去并生成亚胺正离子中间体E,最后,磷亲核试剂对亚胺正离子中间体E进行亲核进攻生成目标产物。

图14 钴催化三级胺sp3C-H磷酰化反应机理
5 钼催化sp3C-H键活化构筑sp3C-P键反应
2012年,Prabhu小组报道了N-芳基四氢异喹啉类化合物和亚磷酸酯类化合物的脱氢氧化偶联反应,在温和反应条件下,以三氧化钼为催化剂,氧气作为氧化剂,实现了N-芳基四氢异喹啉sp3C-H磷酰化,合成了官能团化的α-氨基磷酸酯类化合物(图15)[14]。 虽然N-芳基四氢异喹啉类底物适用范围很广,但只有亚磷酸酯类化合物适合作为反应磷源。在反应机理研究中,该小组认为反应同样是经历一个亚胺正离子中间体的反应历程。该反应体系的亮点在于选择丰度大、清洁的氧气作为体系的氧化剂,符合绿色合成化学的发展。

图15 钼催化N-芳基四氢异喹啉sp3C-H磷酰化反应
6 其他过渡金属催化sp3C-H键活化构筑sp3C-P键反应
2012年,朱成建小组报道了以空气为氧化剂,金络合物为催化剂实现了N-芳基四氢异喹啉的sp3C-H氧化脱氢磷酰化反应,以优秀的收率合成了官能团化α-氨基磷酸酯化合物(图16)[15]。该反应条件温和,底物适用范围广,磷源能同时兼容亚磷酸酯和二苯基磷氧氢。值得注意的是,催化剂金配合物中的配体部分对反应收率起到关键的作用,主要是利用配体去调节催化剂中心金的电性。该小组基于反应机理探究实验,提出反应可能经历一个亚胺正离子的反应历程。
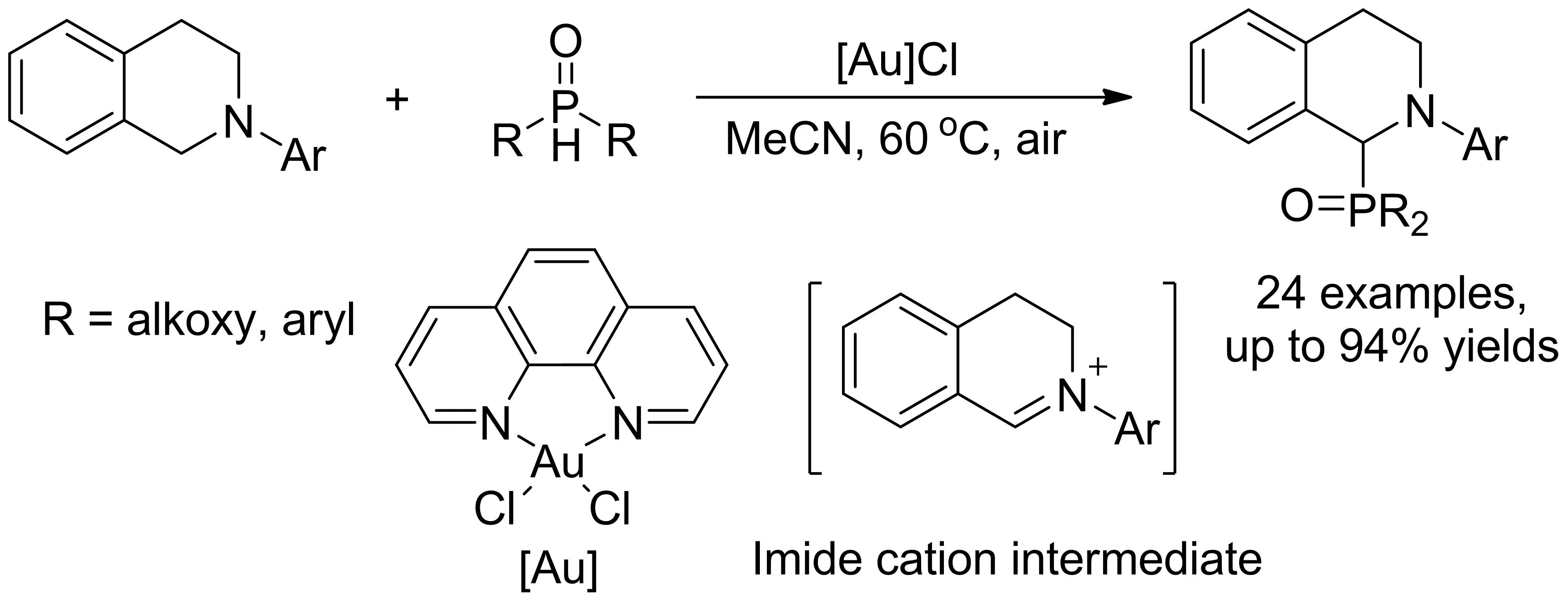
图16 金催化N-芳基四氢异喹啉sp3C-H磷酰化反应
2017年,杨尚东小组报道了铑/镍催化烯丙胺异构化-氢磷酰化反应,通过两种不同过渡金属构成的催化体系催化,实现了烯丙胺底物烯丙位sp3C-H磷酰化反应,合成一系列α-氨基磷酸酯化合物(图17)[16]。文中铑催化体系对烯基胺底物适用范围极广,但不适用于亚磷酸酯类磷源,镍催化体系虽能同时兼容亚磷酸酯和二芳基膦氧氢类磷源,但对烯基胺底物使用范围较窄,两种催化体系互相补充,相得益彰。该小组在机理研究实验中发现,两种催化体系都是通过烯烃异构化形成烯胺中间体的反应历程,且磷源在反应体系中不但是反应底物,同时还起到配体的作用。
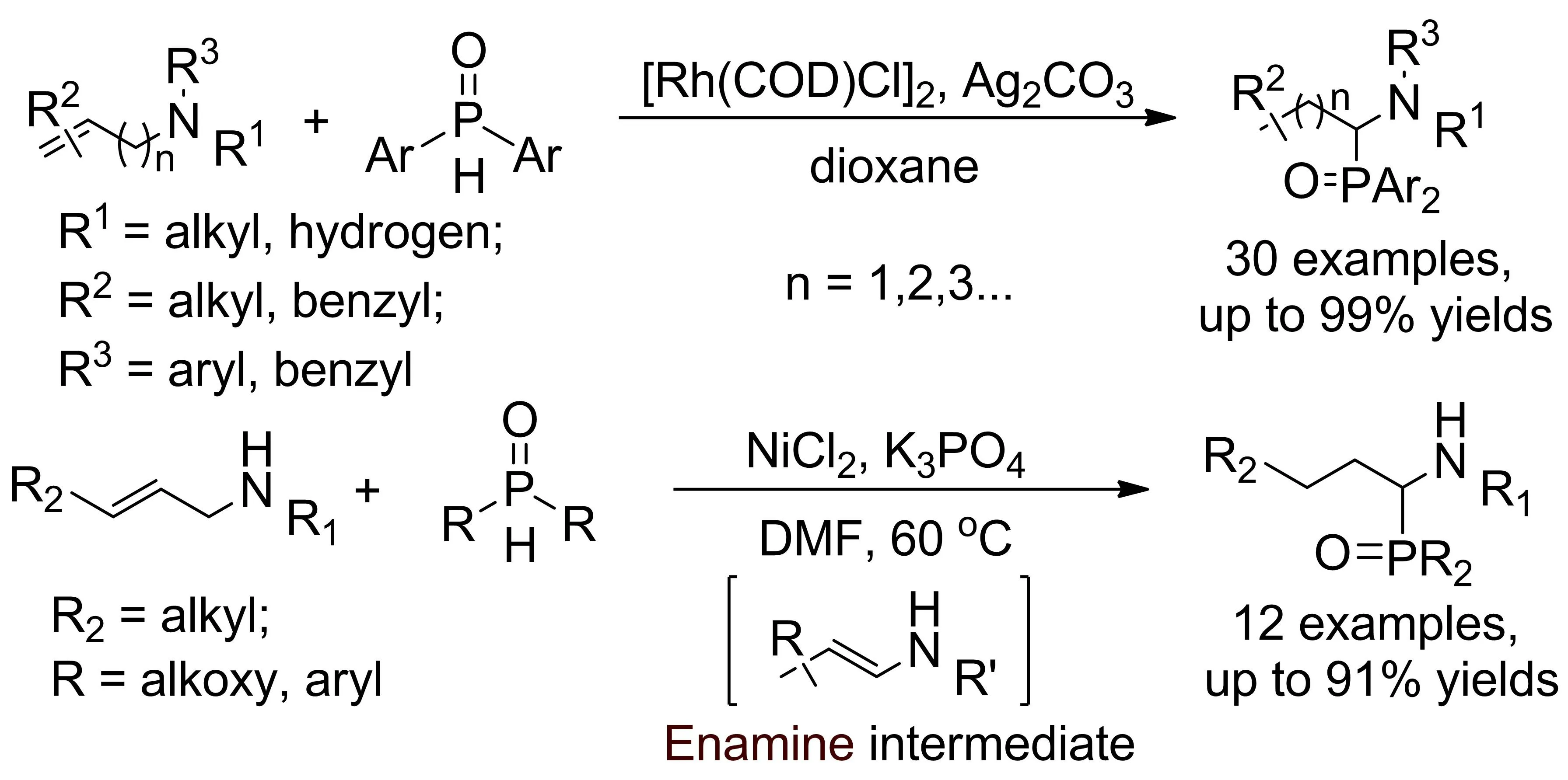
图17 铑/镍催化烯丙胺sp3C-H磷酰化反应
7 总结展望
综上所述,过渡金属催化sp3C-H键活化有效构建sp3C-P键,是合成α-氨基磷酸酯的一种便捷方法。很多结构复杂、官能团化的α-氨基磷酸酯的合成为复杂药物中间体和生物活性分子的合成提供了一种可能的合成策略。通过过渡金属催化sp3C-H键活化构建sp3C-P键,因其具有良好的原子经济性、化学区域选择性和环境友好等优势已取得了重要的进展。但是,过渡金属催化sp3C-H键活化构建sp3C-P键反应仍然存在较多问题,如想在此领域取得突破需要从以下几个方面入手:(1)该类反应底物多数为含氮的胺类底物,底物拓展空间大;(2)目前采用的磷酰化试剂较多集中在价格较高且不太稳定的磷氢试剂,寻找到原料来源广、成本低、稳定的新型绿色磷试剂和开发可回收、高选择性及反应活性的廉价催化剂是未来研究的方向;(3)结合有机光催化反应等新兴技术手段,将光催化、清洁氧化剂氧化应用于此类反应也将是一个巨大机遇和挑战。
猜你喜欢
——碳正离子的产生及稳定性比较
