多组分有机质作用下碳酸钙的矿化现象
2020-06-05王旭辉董发勤李琼芳于璐嘉
王旭辉,董发勤,李琼芳,潘 玲,宋 娜,于璐嘉
(1.西南科技大学生命科学与工程学院,四川绵阳 621010;2.西南科技大学环境与资源学院,四川绵阳 621010;3.西南科技大学固体废物处理与资源化教育部重点实验室,四川绵阳 621010)
黄龙处于高寒地区(平均海拔3 300 m,年平均温度7℃),其独特的钙华沉积方式引起了众多研究者的兴趣。与地质上的矿化作用不同,黄龙钙华的无机相结晶被生物体所分泌的有机质严格控制。有机质可以通过有机-无机界面的分子识别和协同作用(Sheng et al.,2003)选择性地与无机相发生相互作用,从而对矿物的生长、取向、结构和形貌进行调控(Dmitrovic et al.,2012)。矿化过程中,有机质的预组织是生物矿化的模板前提,Oaki等(2012)利用有机酸诱导空心锥状碳酸盐晶体的预组织,氨基酸则能作为诱导CaCO3成核的有机模板(Hu et al.,2010)。当一个有预组织的沉积环境形成时,CaCO3在有机-无机界面处成核并开始生长,其生长取向及速度受有机质控制。Ping等(2018)利用具有有机质特性的chisifica调控纳米球霰石由中心向边缘径向生长,共聚羧酸则能使方解石生长速率降低92.7%(Chhim et al.,2017)。此外,在无外力作用下,CaCO3晶体一般会自主地由亚稳定相转化为热力学最稳定的方解石结构,而通过改变有机质含量能够有效调控晶体结构和形态。乙酸和天冬氨酸均能抑制球霰石向方解石转化,通过改变天冬氨酸的浓度,能够调节球霰石的形状和大小(Tong et al.,2004;Li et al.,2018)。Yang等(2008)利用分子动力学模拟计算了几种低聚糖在方解石晶面上吸附的寡糖单元(半乳糖、甘露糖、鼠李糖、木糖等)含量,发现各糖类在不同晶面上被有选择性地吸附,据此可以预测晶体的形貌。
黄龙水体中生物多样性丰富(李琼芳等,2015)。李骐言等(2013,2014)曾从中分离出2株具有高产碳酸酐酶(CA)活力的嗜冷型菌株,并从其胞外组分中发现了7种有机酸,其中琥珀酸和柠檬酸能够抑制碳酸钙的沉积。嗜冷型菌株分泌的碳酸酐酶能在一定程度上调控碳酸钙的形貌(张文静等,2016),其分泌的核糖及半胱氨酸不仅能促进碳酸钙的沉积,还能诱导文石的产生(陈超等,2017;于璐嘉等,2018)。除了土著细菌,张存凯(2017)从黄龙水体中检测出88种藻类,作为优势藻类,硅藻和黄藻既能为碳酸钙的沉积提供模板,其分泌的有机质对碳酸钙的成核也具有诱导作用。目前的研究多集中于单一有机质对无机矿物的调控方面,然而环境中的生物有机质复杂多样,其与无机离子之间的相互作用也非常复杂,单组分有机质对钙化过程的调控不足以完全揭示黄龙的生物矿化作用,多种有机质对矿物相成核及生长的调控作用仍需深入挖掘。因此,本文依据黄龙水体中嗜冷细菌的胞外产物进行室内模拟研究,建立多种有机酸、氨基酸和单糖共存的混合体系,监测其钙化动力学过程,观察产物形貌和结构,探究参与矿化的主要组分,欲揭示混合体系中有机质对碳酸钙矿化过程的影响机理,这对于探究环境中的协同矿化作用具有重要指导意义。
1 材料和方法
1.1 母液配制
试剂有 CaCl2、NaHCO3、柠檬酸、琥珀酸、L-酪氨酸、L-苯丙氨酸、L-半胱氨酸、D-葡萄糖和 D-核糖等,均为分析纯。
预先按照表1中的有机质成分及浓度配制4种混合母液各1 L,依次命名为A-M、A-O、M-O和A-OM(A为氨基酸,M为单糖,O为有机酸),再配制0.03 M CaCl2溶液和0.07 M NaHCO3溶液各1 L,备用。

表1 母液的成分及浓度 mMTable 1 Composition and concentration of solution
1.2 多组分体系的设置
体系溶液设置为60 mL,其中Ca2+和HCO-3的最终浓度参照黄龙水质分析结果(李骐言,2014)分别设为8.25 mM和16.39 mM。整个反应在静置的培养瓶(300 mL)中进行,将洁净盖玻片置于瓶底以收集沉积物,瓶口用封口膜密封,使用规格10 mL的注射器取样。
实验组:4种混合母液各取30 mL分装至4个培养瓶,每个培养瓶加入15 mL NaHCO3母液及15 mL CaCl2母液,分别命名为 A-M、A-O、M-O和A-O-M。
对照组:移取30 mL超纯水至培养瓶,加入15 mL NaHCO3溶液和15 mL CaCl2溶液。
1.3 矿化动力学监测
采用台式数字酸度计(BANTE PHS-3CW),分别于第1、3、5、7、9 d测试各体系 pH 值,并用注射器取5 mL样品于离心管,抽滤后采用等离子体发射光谱仪(ICP,ThermoFisher iCAP6500)检测Ca2+的质量浓度,波长范围166~847 nm,CID检测器,制冷温度<-40℃。通过指数衰减方程ρ(Ca2+)=ekt+b拟合数据拟合体系的沉积动力学过程,式中,ρ(Ca2+)为Ca2+的质量浓度(mg/L),t为时间(d),k为钙化速率常数,b为截距。d[ρ(Ca2+)]/dt用来计算钙化速率(mg/d)。
1.4 沉积物相和形态的表征
沉淀物在80℃的烘箱里干燥至恒重,并研磨成粉末。通过X射线衍射仪(XRD,X Pert pro,PANalytical B.V.)表征沉积物的结构特征,2θ角3°~80°,管电压 40 kV,管电流 40 mA,扫描步宽 0.02°/min;使用Jade 6.0比对晶体物相;采用傅里叶变换红外光谱仪(FT-IR,Spectrum One,PE)分析化学结构,光谱范围4 000 ~400 cm-1,信噪比100 000∶1,分辨率4 cm-1,DTGS检测器;采用扫描电镜(SEM,UItra55,Carl zeissNTSGmbH)观察沉积物的形貌特征,加速电压25 kV,探针电流200 pA。
2 结果与讨论
2.1 CaCO3的动力学过程
实验通过监测Ca2+浓度和pH值来计算沉积速率,进而比较各体系碳酸钙的沉积情况。结果显示,CaCO3沉积的动力学特征因各体系组分的差异而不同。如图1a所示,各体系的Ca2+浓度在前3 d内均随时间急剧下降,后趋于平缓,但变化趋势以及最终浓度存在差异。在A-M体系中,Ca2+浓度的下降趋势最明显,并始终低于其它体系,沉积速率较对照组提高了36%。M-O、A-O和A-O-M体系中Ca2+浓度变化趋势略为平缓,最终浓度均高于对照组,钙化速率较对照组分别降低了33%、29%、17%(图1b),说明氨基酸与多糖促进了CaCO3的沉积,而有机酸则具有抑制作用。同时,由于在CaCO3的沉积过程中会消耗体系中的HCO-3使得体系溶液的pH值升高(图1c),相比于M-O、A-O和A-O-M体系,A-M体系pH值变化较快,由此也证明A-M体系沉积速率最快。
图1 各体系中碳酸钙的沉积动力学特征Fig.1 Sedimentary kinetics of CaCO3 in various systems during deposition
通过分析实验结果,认为A-M体系促进CaCO3沉积主要有两个原因:①羧基和羟基为CaCO3的沉积提供了成核位点(Cui et al.,2008);② 氨基酸的pI值与pH值不同时会带上电荷,导致氨基酸与Ca2+和CO23-之间发生电荷吸引(陈昕等,2011)。酪氨酸(pI=5.68)、半胱氨酸(pI=5.05)、苯丙氨酸(pI=5.48)的等电点均低于体系pH值,氨基酸释放质子呈负电,与溶液中的正电荷离子相互吸引,在它们相互靠近的过程中,由于溶液中的氨基酸带有大量的羧基,C=O的负电荷在向O移动时会形成强电场,电场对Ca2+具有强烈的吸引力(Jiang et al.,2017),导致 Ca2+的聚集,形成局部过饱和的微环境,因此提高了成核速率(于福家等,2011)。此外,糖类添加剂所携带的大量—OH与Ca2+结合会降低成核活化能(马洁等,2005),能有效协同氨基酸进一步促进晶体成核。
相较于 A-M体系,M-O、A-O、A-O-M体系则抑制了CaCO3的沉积,分析原因可能是有机酸调控了钙化过程。李骐言等(2013)认为有机酸的加入会使体系形成酸性环境,从而抑制HCO-3的解离,因此Ca(HCO3)2则成为部分Ca2+的存在形式。此外,有机酸所携带的羧基在与Ca2+形成螯合物的同时,也降低了Ca2+的饱和度(张群等,2012)。由此可以认为,有机酸对Ca2+的消耗导致了CaCO3沉积量的减少,同时也减缓了其沉积速率。
2.2 沉积物的结构表征
通过XRD图谱对实验形成的沉淀产物的晶体结构进行了分析,结果发现,除A-M体系外的各体系中形成的CaCO3均为方解石,晶体均以(104)晶面为主,表明多组分体系并不影响晶面的优先生长(图2a)。而A-M体系,除了方解石,在2θ为24.9°和27.0°处存在球霰石的衍射峰,分别对应球霰石的(100)和(101)晶面(黄文艺等,2017;张晓蕾等,2018),由Jade 6.0分析其含量仅为晶体总量的2.48%。红外光谱图中712、875及1 422 cm-1处的吸收峰分别对应方解石CO2-3面内弯曲振动、面外弯曲振动及非对称伸缩振动(Park et al.,2015)(图2b)。
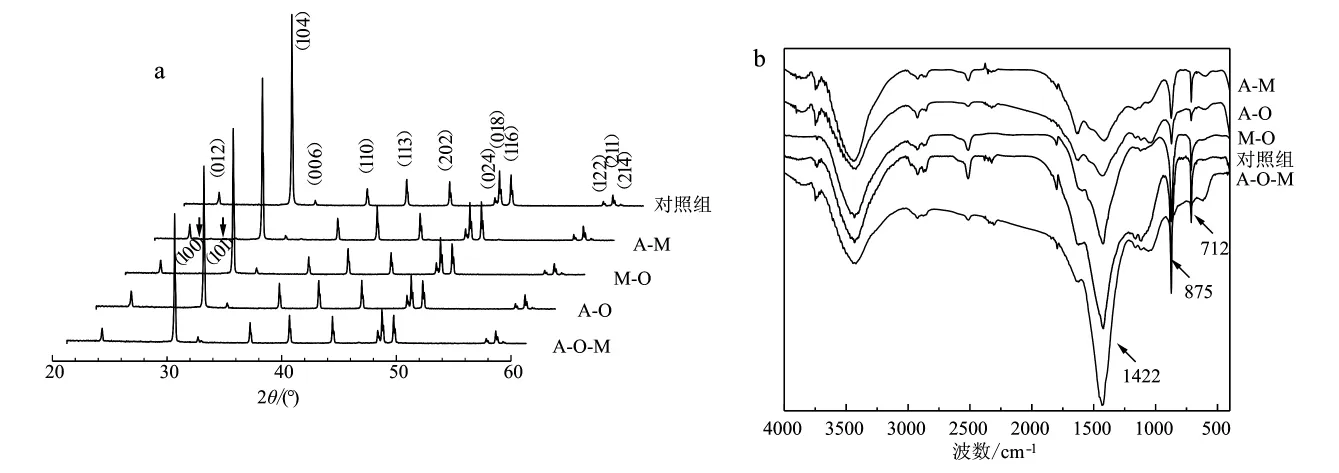
图2 CaCO3的X射线衍射图谱及红外吸收光谱Fig.2 XRD spectra and FT-IR spectra of CaCO3 in various systems
氨基酸和单糖均能诱导球霰石和文石的形成(Jackson and Bischoff,1971;Braissant et al.,2003;李磊等,2017),A-M体系中存在少量的球霰石,说明该体系中存在能诱导球霰石形成的单组分有机质,但M-O、A-O、A-O-M体系的XRD结果显示,多种有机质共同参与钙化时仅形成了方解石一种晶体(图2a),这意味着有机酸抑制了球霰石的合成。此外,本课题组在前期工作中发现了琥珀酸能够诱导球霰石的生成(图3),排除了其对球霰石形成的抑制作用,因此可以推测为柠檬酸调控了CaCO3的晶型。Montanari等(2017)研究发现柠檬酸与Ca2+的比例会影响CaCO3结晶,该比值越高,对球霰石的抑制作用越强,方解石的粒径则越大,并且柠檬酸对文石生长的抑制作用相比于方解石更明显(Gopi et al.,2015)。这与本文实验结果相符合。
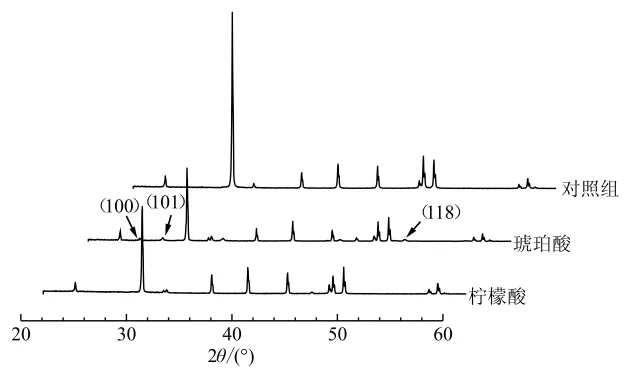
图3 琥珀酸与柠檬酸分别作用下CaCO3的X射线衍射图谱Fig.3 XRD spectra of CaCO3 under the action of succinic acid and citric acid
2.3 沉积物的形态表征
通过观察扫描电镜图谱可以比较各体系对晶体形貌的调控效果。对照组合成了典型的菱面方解石(图4a)。A-M体系合成了粒径为5~10μm的菱面方解石,晶体发育不全,呈台阶状,同时还存在中空环状晶体,粒径约5μm(图4b),结合XRD图谱分析其为球霰石。球霰石含有两个带电荷的钙平面,其电荷密度为6.7 Ca2+/nm2(Lippmann,1973),带负电荷的—COOH引起的强电场有利于与带更强正电荷的晶面相互作用,从而引起结构变化(Tong et al.,2004)。其中空环状的形貌形成过程可能为:①分子间力(如氢键、疏水作用或静电作用)重排分子,形成中空环状结构,亲水端(—OH)朝外,疏水末端(C6H-5)朝内;② 环状结构形成后,Ca2+通过静电作用吸附在亲水性末端,使晶粒沿着环形分子层的表面聚集,形成中空片状晶体(Takahashi et al.,2004)。
M-O、A-O体系均合成长轴径为5~15μm、短轴径为3~5μm的棒状晶体,晶体在基底表面平铺一层,分散性好(图4c、4d)。研究表明,柠檬酸能够强烈抑制CaCO3晶体的生长(Karar et al.,2015),其分子中的羧基可通过配位键与Ca2+形成螯合物(柠檬酸钙),吸附在固-液界面的Ca(OH)2表面,降低晶体的成核速率(伊昌等,2011),导致M-O、A-O体系合成的晶体较少。其次,柠檬酸α-C上的羟基取代基会选择性地吸附在晶体表面,不仅通过改变晶体表面的能量分布,诱导晶体沿c轴生长(林荣毅等,2002;Westin and Rasmuson,2005),而且形成空间位阻,减弱了晶体之间的吸引力,导致M-O和A-O中的晶体不团聚。
A-O-M体系中的晶体个数多、尺寸小(粒径约为1~3μm),且晶体聚集成簇状(图4e)。与M-O、A-O体系晶体生长方式不同,氨基酸协同单糖提高了A-O-M体系的过饱和度,有效克服了柠檬酸对晶体成核的抑制作用。经典成核理论Gibbs-Thomson公式表明,保持溶液的过饱和微环境可以提高晶体的成核速率(Tong et al.,2004)。当晶体的成核速率较高时,晶体的生长会相对受到抑制,有助于晶体的细化(Yang and Nan,2010)。A-O-M体系中有机质含量最高,既能提供充足的成核位点,还能促进过饱和微环境的形成,以致晶体成核速率被提高,用于晶体生长的离子相对变少,因此形成的晶体粒径小。

图4 CaCO3的扫描电镜图像Fig.4 The SEM images of CaCO3 in various systems
3 结论
本文通过实验比较了多种有机质共同作用下碳酸钙的矿化现象,得到了以下结论:
(1)氨基酸协同单糖促进了CaCO3的沉积,并延长了中空的环状球霰石的稳定时间;
(2)当氨基酸或单糖单独与有机酸混合参与矿化时,钙化速率大幅度降低,晶体的结构与形貌受柠檬酸强烈调控;
(3)增加有机质含量能够有效克服柠檬酸对晶体成核的抑制作用。
