气相色谱-串联质谱法测定2-氰基溴苄中3-氰基溴苄和4-氰基溴苄的含量
2020-06-03庄航
庄 航
(宿迁市食品药品检验所,宿迁223800)
苯甲酸阿格列汀是以3-甲基-6-氯尿嘧啶和2-氰基溴苄为起始原料,在碱性条件下发生烷基化反应得到2-(6-氯-3-甲基-2,4-二氧代-3,4-二氢-2H-嘧啶-1-基甲基)-苄腈,再与(R)-3-氨基哌啶二盐酸发生取代反应得到(R)-2-[(6-(3-氨基哌啶-1-基)-3-甲基-2,4-二氧代-3,4-二氢嘧啶-1(2 H)-基)甲基]苄腈(阿格列汀游离碱),阿格列汀游离碱再与苯甲酸成盐得到苯甲酸阿格列汀[1-5]。由于2-氰基溴苄是合成苯甲酸阿格列汀的关键起始原料[6],其产品的质量直接影响苯甲酸阿格列汀的安全性,需要严格控制。2-氰基溴苄在合成过程中可能会产生少量的同分异构体3-氰基溴苄和4-氰基溴苄副产物[7-8]。2-氰基溴苄、3-氰基溴苄和4-氰基溴苄均为含卤代烷烃类结构的基因毒性杂质,需要对2-氰基溴苄中的3-氰基溴苄和4-氰基溴苄的残留量进行控制,以降低苯甲酸阿格列汀中基因毒性杂质的水平[9-12]。参考ICH M7基因毒性杂质评估和控制指导原则中潜在基因毒性杂质限度制定依据,以毒理学关注阈值(TTC,1.5μg·d-1)法计算[10],苯甲酸阿格列汀的最大日剂量为25 mg,2-氰基溴苄中同类基因毒性杂质所允许的最大残留量为60 mg·L-1。
经查询,国内外尚无2-氰基溴苄、3-氰基溴苄和4-氰基溴苄的分离检测方法的研究报道。本工作采用气相色谱-串联质谱法(GC-MS/MS)结合中等极性的毛细管色谱柱同时分离了上述3种化学性质相似的同分异构体极性化合物,可快速准确测定2-氰基溴苄中潜在基因毒性杂质3-氰基溴苄和4-氰基溴苄的残留量,以期为苯甲酸阿格列汀的用药安全提供保障。
1 试验部分
1.1 仪器与试剂
安捷伦7890B-7000C型气相色谱-串联质谱联用仪,配Mass Hunter工作站软件,XPE56DR型电子天平。
3-氰基溴苄和4-氰基溴苄混合标准溶液:分别称取3-氰基溴苄和4-氰基溴苄标准品各10 mg,用乙腈溶解并转移至10 mL容量瓶中,用乙腈定容,混合均匀,配制成1 000 mg·L-1的混合标准储备溶液;移取混合标准储备溶液100μL于10 mL容量瓶中,用乙腈稀释至刻度,混合摇匀,配制成10 mg·L-1的混合标准溶液;再移取10 mg·L-1的混合标准溶液100μL于10 mL容量瓶中,用乙腈稀释至刻度,混合摇匀,配制成100μg·L-1的3-氰基溴苄和4-氰基溴苄的混合标准溶液。
系统适用性溶液:称取试样约25 mg,用乙腈溶解并转移至25 mL容量瓶中,用乙腈定容,混合均匀后,移取1 mL于10 mL容量瓶中,加入100μg·L-1的3-氰基溴苄和4-氰基溴苄的混合标准溶液0.3 mL,用乙腈稀释至刻度,混合均匀。
空白溶液:乙腈。
3-氰基溴苄和4-氰基溴苄标准品的纯度均为98.86%;待测样品中2-氰基溴苄的纯度不小于99%;乙腈为色谱级。
1.2 仪器工作条件
1)色谱条件 以50%苯基-50%二甲基聚硅氧烷为固定液的VF-17MS毛细管色谱柱(30 m×0.25 mm,0.25μm);进样口温度为260℃;载气为氦气(纯度为99.999%),流量为1.5 mL·min-1;碰撞气为氮气(纯度为99.999%),流量为1.5 mL·min-1;淬灭气为氦气(纯度为99.999%),流量为2.25 mL·min-1;进样量为 1μL;衬管为 5190-2295型分流衬管;进样口为多模式进样口;分流进样,分流比为5∶1。柱升温程序:初始温度为60℃;以20℃·min-1的速率升温至180℃;再以5℃·min-1的速率升温至185℃,保持2 min;最后再以40℃·min-1的速率升温至300℃,保持2 min。
2)质谱条件 电子轰击离子源(EI);多反应监测(MRM)模式;氰基溴苄的定性离子对的质荷比m/z为116/63,116/116,碰撞能量分别为35,10 e V;定量离子对的质荷比m/z为116/89,碰撞能量为15 e V。
1.3 试验方法
称取待测样品25 mg,用乙腈溶解并转移至50 mL容量瓶中,用乙腈定容,混合均匀,得到待测样品溶液。移取待测溶液1μL,按照仪器工作条件对其进行测定。
2 结果与讨论
2.1 色谱条件的选择
2.1.1 色谱柱
3种氰基溴苄均为沸点较高的极性化合物,试验首先考虑采用弱极性的HP-5毛细管色谱柱(30 m×0.320 mm,0.25μm)对2-氰基溴苄、3-氰基溴苄和4-氰基溴苄进行分离,为了缩短分析时间,色谱柱升温程序设定为:初始温度为60℃,维持3 min,再以20℃·min-1的速率升温至170℃,保持5 min。载气流量为1.5 mL·min-1,分流进样,分流比为20∶1。向待测样品中加入3-氰基溴苄和4-氰基溴苄的混合标准溶液,按照上述仪器工作条件对其进行测定,色谱图见图1。
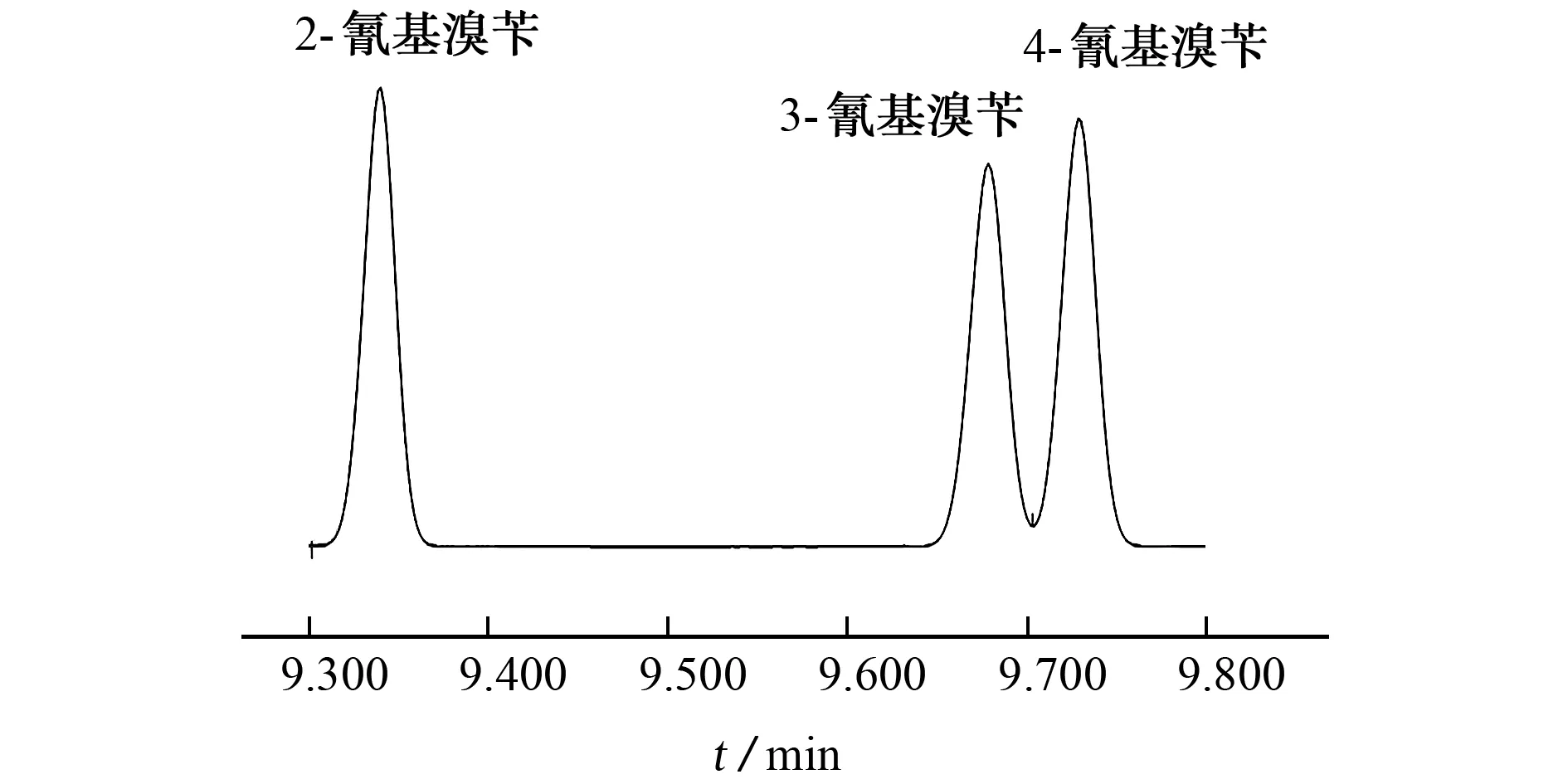
图1 2-氰基溴苄、3-氰基溴苄和4-氰基溴苄在HP-5毛细管色谱柱下的分离色谱图Fig.1 Separation chromatogram of 2-cyanobenzyl bromide,3-cyanobenzyl bromide and 4-cyanobenzyl bromide under HP-5 capillary column
结果表明:4-氰基溴苄和3-氰基溴苄对应的相邻色谱峰的分离度为1.35,小于1.5,说明未实现基线分离。
试验进一步采用DB-5HT毛细管色谱柱(30 m×0.250 mm,0.1μm)对2-氰基溴苄、3-氰基溴苄和4-氰基溴苄进行分离,该色谱柱经交联、键合,对高沸点组分可提供更快速的洗脱效率和更好的分离效果。色谱柱升温程序设定为:初始温度为45℃,保持3 min;以20℃·min-1的速率升温至120℃,保持5 min;再以30℃·min-1的速率升温至210℃,保持1 min。按照上述仪器工作条件对2-氰基溴苄、3-氰基溴苄和4-氰基溴苄的混合标准溶液和供试品溶液进行测定,结果表明:3-氰基溴苄和4-氰基溴苄对应的相邻色谱峰的分离度提高至1.9,大于1.5,实现了基线分离,但是在2-氰基溴苄供试品溶液中4-氰基溴苄和3-氰基溴苄出峰与2-氰基溴苄部分重合,导致该色谱条件下,4-氰基溴苄和3-氰基溴苄的回收率高于150%。
考虑到2-氰基溴苄、3-氰基溴苄和4-氰基溴苄均为含氰基极性基团的化合物,试验选择将弱极性毛细管柱更换为中等极性固定液的毛细管柱,以增加固定液与三者的偶极作用。以VF-17MS毛细管色谱柱(30 m×0.25 mm,0.25μm)为分析柱,在1.2节处的柱升温程序下进行测定,结果显示3-氰基溴苄和4-氰基溴苄的分离度可达2.8,表明VF-17MS分离效果高于另外两种弱极性毛细管柱,色谱图见图2。
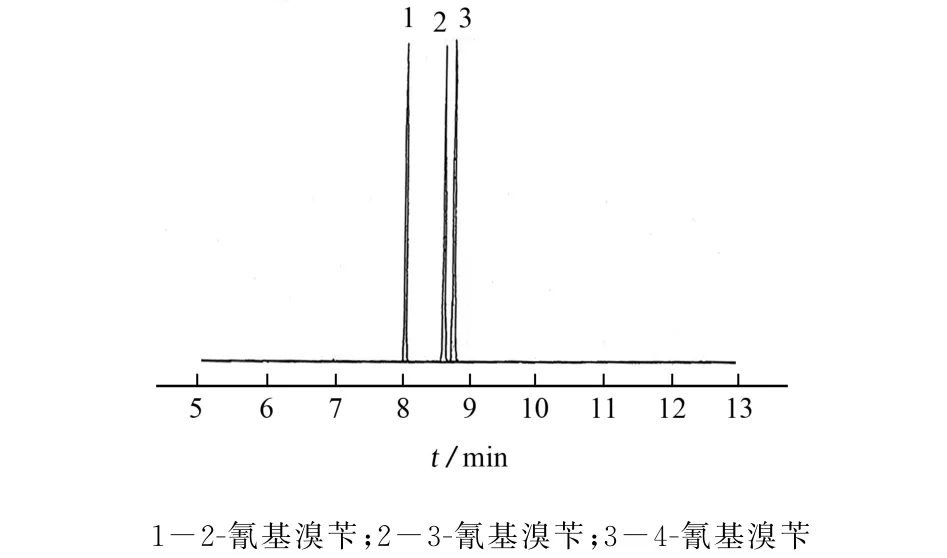
图2 2-氰基溴苄、3-氰基溴苄和4-氰基溴苄在VF-17MS毛细管色谱柱下的分离色谱图Fig.2 Separation chromatogram of 2-cyanobenzyl bromide,3-cyanobenzyl bromide and 4-cyanobenzyl bromide under VF-17MS capillary column
2.1.2 柱温
以VF-17MS毛细管色谱柱(30 m×0.25 mm,0.25μm)为分析柱,在0~20 min内,色谱柱的温度先由50℃升温至300℃,2-氰基溴苄、3-氰基溴苄、4-氰基溴苄的出峰时间分别为8.126,8.451,8.527 min(见图3,图3中柱升温程序A:0~20 min内,温度由50℃升温至300℃;柱升温程序B:初始温度60℃,以20℃·min-1的速率升温至180℃;再以5℃·min-1的速率升温至185℃,保持2 min;最后以40℃·min-1的速率升温至300℃,保持2 min)。VF-17MS毛细管色谱柱的死时间为1.470 min,扣除死时间后得到化合物的调整保留时间分别为6.656,6.981,7.057 min,该时间对应的柱温箱温度即为该化合物的特征流出温度,2-氰基溴苄、3-氰基溴苄、4-氰基溴苄的特征流出温度分别为183.12,189.62,191.14℃。因为气相色谱仪采用气浴加热方式,柱温箱温度会略微高于色谱柱实际温度,且升温速率越大该差值越大[13-15]。当以20℃·min-1的升温速率加热时,需要将柱温箱温度扣减5℃得到色谱柱温度,2-氰基溴苄的流出温度大致为180℃,3-氰基溴苄和4-氰基溴苄的流出温度约为185℃。试验中采用快速升温的方式升温至180℃使得2-氰基溴苄流出,后缓慢升温至185℃,在该温度下恒温一倍的色谱柱死时间以提高3-氰基溴苄和4-氰基溴苄的分离度,最后快速升温至300℃,老化色谱柱。
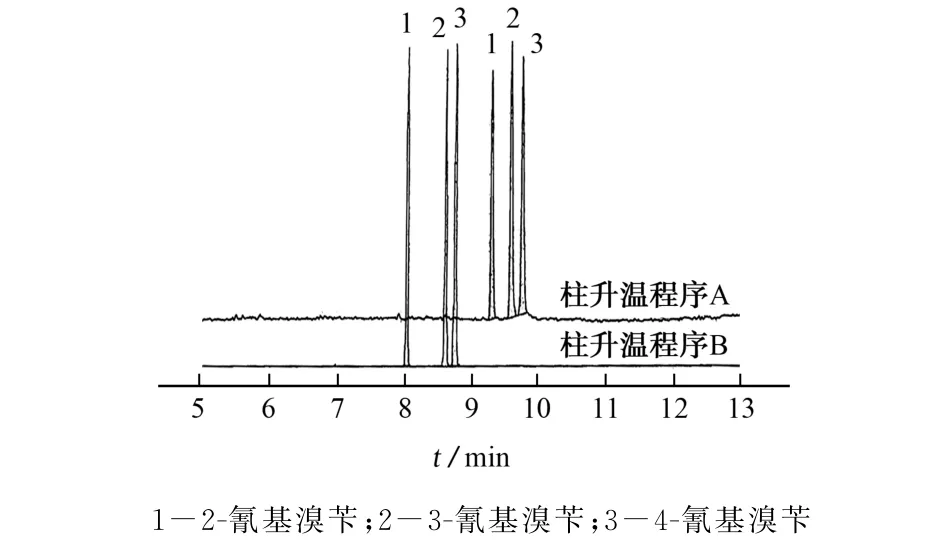
图3 2-氰基溴苄、3-氰基溴苄和4-氰基溴苄在不同的色谱柱升温程序下的分离色谱图Fig.3 Separation chromatogram of 2-cyanobromobenzyl,3-cyanobromobenzyl and 4-cyanobromobenzyl under different chromatographic column heating procedure
2.2 质谱条件的选择
2-氰基溴苄、3-氰基溴苄和4-氰基溴苄的质谱基峰均为116,根据结构式可知碎片均来自于氰苄基。采用子离子扫描方式,选择质荷比(m/z)116作为初级离子,碰撞能量设置为10,15,25,35 eV,子离子扫描m/z范围为50~150。分析质谱子离子扫描数据显示m/z116能得到m/z为89和63两个碎片离子,同时未打碎的m/z116处也有较强响应。由结果得知最佳信噪比离子对的m/z为116/89,选择该离子对为定量离子对;m/z为116/63和m/z116/116离子对作为定性离子对。2-氰基溴苄、3-氰基溴苄和4-氰基溴苄的最佳碰撞能量分别为15,35,10 e V,且3种化合物质谱信息一致。
2.3 标准曲线、检出限和测定下限
配制质量浓度分别为5,15,22.5,30μg·L-1的3-氰基溴苄和4-氰基溴苄的混合标准溶液系列,按照仪器工作条件进行测定,以3-氰基溴苄和4-氰基溴苄的质量浓度为横坐标(x),与其对应的峰面积为纵坐标(y)绘制标准曲线。结果表明:3-氰基溴苄和4-氰基溴苄的质量浓度均在5.0~30.0μg·L-1内与其对应的色谱峰面积之间呈线性关系,3-氰基溴苄的线性回归方程为y=2.996×103x-2.693×103,相关系数为0.999 8,y轴截距比100%响应值为6.3%;4-氰基溴苄的线性回归方程为y=2.659×103x-3.710×103,相关系数为0.998 7,y轴截距比100%响应值为10%。
以3倍信噪比计算方法的检出限(3S/N),以10倍的信噪比计算方法的测定下限(10S/N)。结果表明:3-氰基溴苄和4-氰基溴苄的检出限均为2.0μg·L-1,测定下限均为5.0μg·L-1。
2.4 精密度试验
平行配制6份系统适用性溶液,按仪器工作条件进行测定,计算3-氰基溴苄和4-氰基溴苄测定值的相对标准偏差(RSD)。结果发现:3-氰基溴苄测定值的RSD为1.2%,4-氰基溴苄测定值的RSD为3.3%,说明该方法重复性良好。
由不同的分析人员在不同的日期使用不同仪器按照上述方法进行试验。结果发现:12份待测溶液中3-氰基溴苄测定值的RSD为4.9%,4-氰基溴苄测定值的RSD为3.5%。表明该方法的再现性良好。
2.5 回收率试验
按照试验方法对阴性样品进行低、中和高等3种不同浓度水平的加标回收试验,计算3-氰基溴苄和4-氰基溴苄的回收率,结果见表2。
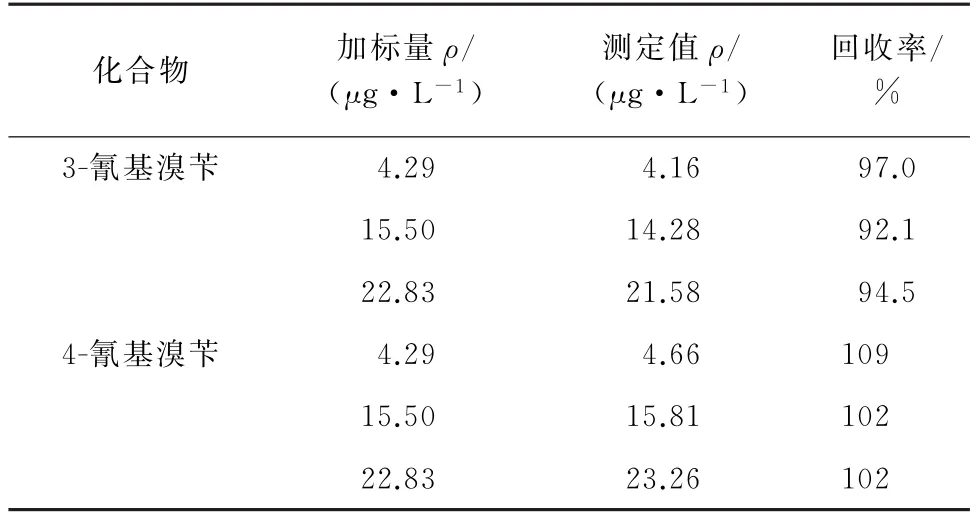
表1 回收试验结果Tab.1 Results of test for precision
由表2可知:3-氰基溴苄和4-氰基溴苄的回收率分别为92.1%~97.0%和102%~109%。
2.6 溶液的稳定性试验
试验考察了待测样品溶液、系统适用性溶液以及3-氰基溴苄和4-氰基溴苄标准溶液在25℃条件下放置0,2,5,9,12,18 h后的峰面积变化情况。结果表明:3-氰基溴苄和4-氰基溴苄标准溶液、待测样品溶液及系统适用性溶液在25℃条件下放置18 h后,3-氰基溴苄和4-氰基溴苄的峰面积与放置0时相比,峰面积的相对偏差小于5%。表明各溶液在25℃条件下放置18 h后稳定性良好。
2.7 专属性试验
移取1.1节处的各溶液1μL注入气相色谱-串联质谱联用仪,记录其色谱图。结果表明:空白溶液不干扰3-氰基溴苄和4-氰基溴苄测定,2-氰基溴苄、3-氰基溴苄和4-氰基溴苄保留时间分别为8.058,8.617,8.759 min,相邻色谱峰的分离度分别为7.1,2.8。表明该方法专属性良好。
2.8 耐用性试验
试验通过改变载气流量为(1.1±0.1)mL·min-1、分流比为4∶1~6∶1、电子轰击能量变化范围为-5~+5 e V来评估试验条件有微小变化时,3-氰基溴苄和4-氰基溴苄测定结果的受影响程度。系统适用性试验中,电子轰击能量变化范围为-5~+5 eV,分流比分别变为4∶1及6∶1后,3-氰基溴苄和4-氰基溴苄测定值的RSD均小于5.0%;但是流量变化±0.1 mL·min-1后,3-氰基溴苄和4-氰基溴苄测定值的RSD均大于5.0%,提示在选定的色谱条件下载气的流量需准确设定。
2.9 样品分析
按照试验方法对批号分别为ALOB1710009、ALOB1810007和ALOB1902002的3批样品进行测定。结果表明:3批样品中均未检出3-氰基溴苄和4-氰基溴苄。
本工作采用GC-MS/MS结合中等极性的毛细管色谱柱同时分离测定了2-氰基溴苄中2种化学性质相似的含卤代烷烃类基因毒性结构的同分异构体极性化合物,方法操作简便、分离度好,能快速测定2-氰基溴苄中潜在基因毒性杂质3-氰基溴苄和4-氰基溴苄的微量残留,可为苯甲酸阿格列汀的用药安全提供保障。
