磷酸蒸馏-离子色谱法测定药物中的氰根总量
2020-06-03栾绍嵘刘鹏宇张芳芳SOLANGEMuhayimana倪力军
栾绍嵘,刘鹏宇,张芳芳,SOLANGE Muhayimana,倪力军∗
(1.华东理工大学 化学与分子工程学院,上海200237; 2.赛默飞世尔科技(中国)有限公司,上海201203;3.华东理工大学 药学院,上海200237)
氰化物作为剧毒物质,不仅对人类的生理系统产生极端的毒性[1],而且在冶金、医药、电镀、橡胶和农药等领域所引起的环境问题也日益严重[2-6]。许多药物在合成过程中需要用到含氰化物的原料,或者工艺生产过程中会产生少量氰化物,为了对药物中的氰化物进行监控,避免对人类的身体健康造成危害,亟需建立一种快速、简便的测定药物中痕量氰化物的分析方法。
氰化物的检测方法主要有异烟酸-吡唑啉酮分光光度法[7]、气相色谱法[8]、液相色谱法[9]、荧光光谱法[10]、流动注射分析法[11]和极谱法[12]等。中国药典2015版采用紫外分光光度法对氰化物进行半定量测定,美国药典则采用普鲁士蓝法对氰化物进行定性。这些方法均存在干扰多、操作复杂、检出限低、分析成本高以及设备操作要求高等缺点[13]。离子色谱法具有操作简单、快速简便、灵敏度高以及选择性好等优点,是化工、环境、能源、制药行业样品中分析阴、阳离子的重要手段[14-16]。对于电导检测器,氰根离子在抑制器上反应生成弱解离的易挥发的氢氰酸,其电导值低以致无法检测[17]。ROCKLIN[18]首次报道采用电化学检测器和银电极检测氰化物,提高了选择性和灵敏度,并广泛用于食品[19]、废水[20]、饮用水[21-22]、固体废弃物[23]和制药[24]等领域。
目前,复杂基体药物中的基体对氰根的检测存在很大的干扰,导致基线不稳定,氰根的检出限高,达不到制药行业的高要求,这是亟待解决的一个大问题。本工作通过向药物水溶液中加入磷酸,加热蒸馏,将氰根以氰化氢的形式蒸出,经过冷凝并被氢氧化钠溶液吸收后,采用离子色谱-安培检测器法测定溶液中氰根(CN-)的含量,从而得到原药物样品中的氰根含量。该方法有效避免了基体对待测物的影响,灵敏度高,结果满意,能够满足制药行业对药物中氰根的检测要求。
1 试验部分
1.1 仪器与试剂
ICS 6000型离子色谱仪,配AS-AP自动进样器、安培检测器和Chromeleon 7.2色谱工作站;以Ag电极为工作电极,以Ag/AgCl为参比电极;Milli-Q型超纯水机;AL 204型分析天平;YH型电加热器;On Guard RP前处理柱。
氰根标准储备溶液:1 000 mg·L-1,介质为水。
氰根标准溶液系列:分别移取氰根标准储备溶液2.5,5.0,12.5,25μL于25 mL容量瓶中,用水定容,混合均匀,配制成0.1,0.2,0.5,1.0 mg·L-1的氰根标准溶液系列。再移取0.2,0.5 mg·L-1的氰根标准溶液1.0 mL,用水稀释至10.0 mL,配制成0.02,0.05 mg·L-1的氰根标准溶液。
50%(质量分数,下同)氢氧化钠溶液为优级纯;磷酸为分析纯;试验用水为超纯水。
待测药品的主要成分为氯化钠、氯化钾、无水葡萄糖和枸橼酸钠;4批药品的批号为1701、1702、1703和1704,均来自生产企业。
1.2 色谱条件
IonPac AS11-HC阴离子色谱柱(250 mm×4 mm);IonPac AG11-HC阴离子保护柱(50 mm×4 mm);进样量为25μL;流量为1.0 mL·min-1;柱温箱的温度为30℃;流动相为4 mmol·L-1的氢氧化钠溶液,等度洗脱。
1.3 试验方法
称取样品0.5 g于250 mL三口烧瓶中,加入50 mL水,迅速加入1 mL磷酸,立即盖好瓶塞,进行加热蒸馏,所得气体用5 mL的20 mmol·L-1氢氧化钠溶液吸收,将吸收液用水定容至25 mL容量瓶中,得到待测溶液,按照色谱条件进行测定。
2 结果与讨论
2.1 前处理方式的选择
批号为1701的药品样品经过不同前处理方式得到的色谱图见图1。通过与空白溶剂和20 mmol·L-1氢氧化钠溶液溶解后加On Guard RP前处理柱处理后进样后得到的色谱图对比可知:在此条件下,氰根没有受到高盐基体的干扰,噪声低、灵敏度高,能够用于实际复杂基体药物中氰根的分析。两种处理方式所得结果的不同是因为样品中含有大量的氯离子,虽然经On Guard RP前处理柱处理后可以除去大分子有机物基体,但大量氯离子的存在对氰根的测定存在干扰。经过酸蒸后,样品中的氯转化成盐酸,由于盐酸的沸点比氢氰酸高,所以,在95℃下不会被蒸出来,因此不会干扰已蒸出的氰根的测定。
2.2 酸蒸条件的选择
2.2.1 无机酸的种类
无机酸的作用是提供酸性环境,将氰根转化为氰化氢。因此,不能选择挥发性酸,而硫酸和硝酸的酸性太强,不容易控制溶液的酸度。综合考虑,试验选择中等强度酸性的磷酸。
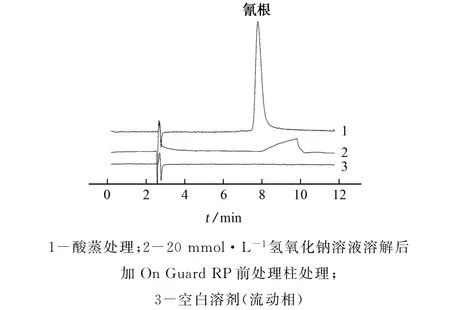
图1 不同前处理方法得到批号为1701样品中氰根的色谱图Fig.1 Chromatograms of CN-in the 1701 sample was obtained by different pretreatment methods
2.2.2 蒸馏温度
试验考察了蒸馏温度分别为80,90,95,100℃时对馏出液速率的影响。结果发现:当蒸馏温度为80,90℃时,由于没有达到水的沸点,水蒸气在甁壁上冷凝,没有冷凝液流出;当蒸馏温度为100℃时,馏出速率较高,不利于氰根的吸收;当蒸馏温度为95℃时,不仅能维持合适的蒸馏速率(2~4 mL·min-1),而且氰根的蒸出和吸收比较完全。
2.2.3 馏出液体积
以批号为1701的药品样品为研究对象,试验考察了馏出体积分别为15,20,30 mL时对氰根回收率的影响,结果见表1。
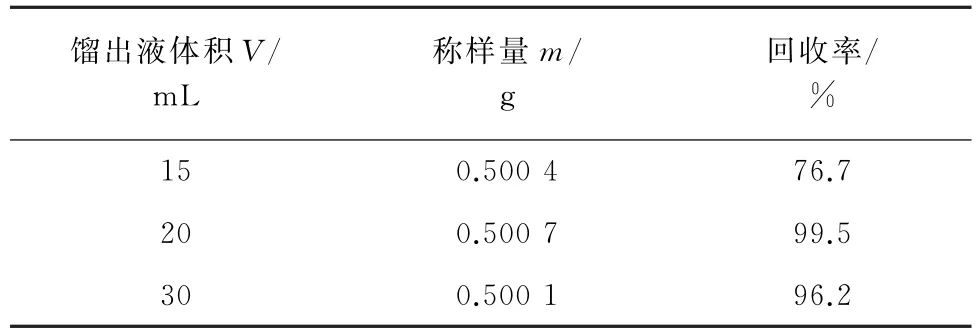
表1 不同馏出液体积下样品中氰根的回收率Tab.1 Recoveries of CN-in the sample under different distillation volumes
由表1可知:当馏出液体积为15 mL时,氰根的回收率较低,未能完全蒸出;当馏出液体积为20 mL和30 mL时,样品溶液中的氰根能够被完全蒸出。从节约时间和成本的角度考虑,试验选择馏出液的体积为20 mL。
2.3 标准曲线、检出限和测定下限
按照色谱条件对氰根标准溶液系列进行测定,以氰根的质量浓度为横坐标,与其对应的峰面积为纵坐标绘制标准曲线。结果表明:氰根的质量浓度在0.02~1.0 mg·L-1内与其对应的峰面积之间呈线性关系,线性回归方程为y=8.028x-0.006,相关系数为0.999 7。
以3倍信噪比和10倍信噪比计算方法的检出限(3S/N)和测定下限(10S/N),结果分别为0.004,0.014 mg·L-1,实际样品的检出限为0.010μg·g-1。
2.4 精密度和回收试验
按照试验方法对实际药品样品进行加标回收试验,每个加标样品平行测定6次,计算回收率和测定值的相对标准偏差(RSD)。精密度和回收试验结果见表2。
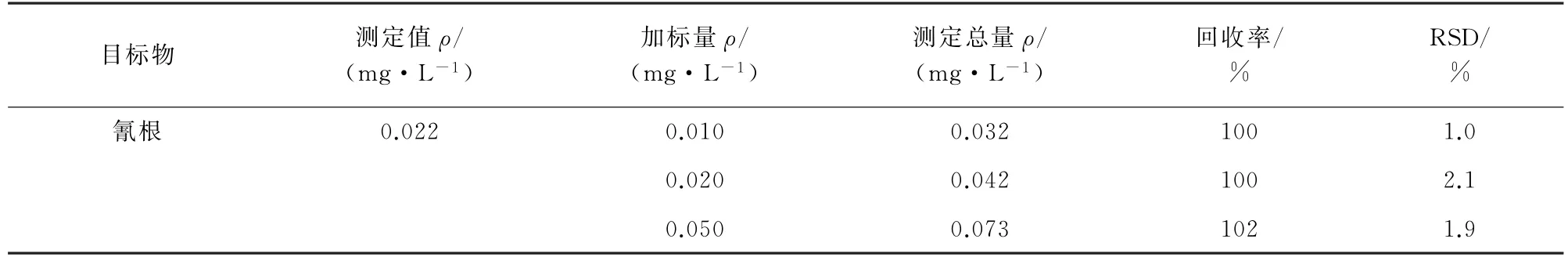
表2 精密度和回收试验结果(n=6)Tab.2 Results of tests for precision and recovery(n=6)
由表2可知:氰根的回收率为100%~102%,测定值的RSD为1.0%~2.1%,表明该方法准确度高,重复性好,能够满足复杂基体药物中氰根的分析需求。
2.5 实际样品的测定
按照试验方法对批号分别为1701、1702、1703、1704的4批药品样品进行测定。结果发现:4批药品样品中氰根的质量分数分别为1.12,9.56,0.105,3.24μg·g-1。
本工作建立了磷酸蒸馏-离子色谱法测定药品样品中氰根总量的方法。药物样品在加入磷酸的条件下,加热蒸馏,氰根以氰化氢的形式被氢氧化钠溶液吸收后,采用离子色谱法进行测定。与中国药典2015版中所采用的3种氰化物检查法相比,该方法灵敏度高、精密度和重复性好,定量准确度高,用于实际样品分析,结果非常满意。磷酸蒸馏和离子色谱法相结合有效解决了复配药物中复杂有机和无机基体的干扰,可为复配药物中氰根的测定提供新方法和新思路。
