基于第一性原理对SmCo 纳米团簇磁性的研究
2020-06-03郑燕飞
郑燕飞
(西南大学物理科学与技术学院,重庆400715)
1 概述
纳米团簇(nanoclusters),是由几个或者上千万个原子或分子通过化学或物理的相互作用力结合在一起形成相对稳定的集合体。有人认为团簇是介于微观粒子以及宏观物质之间的新的物质结构[1]。目前人们对团簇的研究兴趣越来越高涨的部分原因是纳米团簇材料构成了一种新的物质,在材料应用行业有着很大的发展潜能,它的性质既不同于单个的原子、分子,也不同于块体物质。另外还有几个重要的原因是:(1)团簇的性质变化具有量子尺寸效应[2,3,4]。(2)团簇的比表面积大。(3)纳米团簇具有小尺寸效应。其实纳米团簇的结构和稳定性都会随着原子尺寸的变化而发生巨大的变化。
近年来,SmCo 合物的磁性材料器磁性和电性之间有趣而显著的耦合关系而引起了人们的广泛关注。尤其是含稀土纳米团簇的材料方面,具有潜在的重要性。对稀土团簇的研究有利于进一步了解团簇性质的本质,在物理和化学领域都有重要的意义。含稀土元素的纳米团簇结构,由于其尺寸和表面效应引起的新型磁性能特性,正成为科学技术研究的热点。
2 计算方法
本文所有的计算都是采用VASP 程序包进行计算,VASP 程序包是基于密度泛函理论下的平面波赝势展开对于自旋局域密度泛函理论用迭代自洽的方法求解。所有的计算都在Perdew、Burke Ernzerhof (PBE)版本的交换相关电位的广义梯度近似(GGA)下进行的[5]。对于平面波基集,我们使用了动能截止为360eV 的投影增广波(PAW)方法,我们已经使用了这个该软件的程序[6,7]。该团簇被放置在一个体积为30 埃的大型立方体超晶胞中央,在弛豫过程中保持晶格向量不变,保证了周期像之间的充分分离。通过充分结构弛豫设计出平衡原子合理而稳定的构型,在团簇计算中设置每个原子上的受外为0.01 eV/A,总能量收敛标准为10-5eV。为了提高电子自洽场(SCF)的收敛性,将如何确定电子的部分占据数参数,设置使用费米计算ISMEAR=1,设置展开的宽度为0.2 eV,这样可以使得含有稀土纳米团簇的收敛性更好点。
3 结论与讨论
3.1 团簇结构分析
本文研究的纳米团簇结构为典型的SmCo 组成的笼状球形团簇,中间一个Sm 原子被周围6 个Co 原子包围,结构为层状叠加高对称球状纳米团簇结构,如图(1)所示:
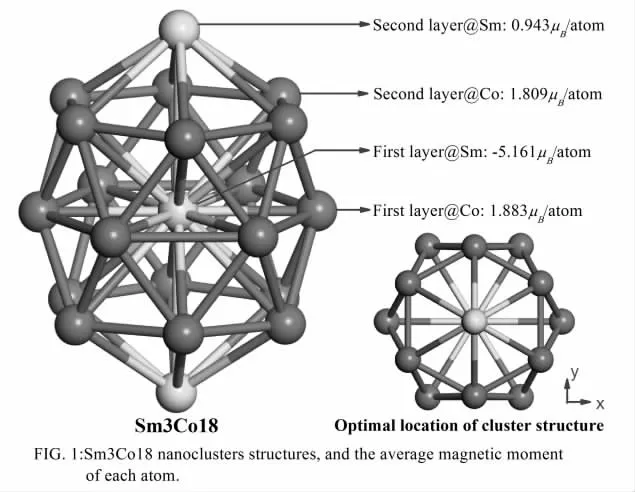
图1
为了研究方便定义中间的Sm 原子为第一层Sm,而中间的6 个Co 原子组成的六边形为第一层Co,本结构为高对称结构,对称性为D6H, 对称主轴为Z 轴,与3 个Sm 原子组成的线共线,从而合理的设计出本文的研究纳米团簇结构,使用VASP 软件进行结构优化,摆放的位置如图1 右下方所示,通过计算发现中间的Sm 原子为反铁磁性,磁性最大,而该团簇结构两端处为铁磁性的Sm 原子,磁性最小。这样设计出的纳米团簇结构有很大的磁性,是新型高磁性纳米复合材料。
3.2 团簇磁性的分析
如图2 所示的为Sm3Co18 纳米团簇中每个原子的平均轨道磁矩、平均自旋磁矩和通过LS 耦合形成的平均磁矩。通过vasp 计算团簇的SOC,设置磁化方向沿x 方向和y 方向,通过计算发现x 方向比y 方向的能量低,因此该纳米团簇结构的易磁化轴为x,取易磁化方向的自旋磁矩和轨道磁矩进行磁性分析,从图2 知,第一层的Sm 原子的自旋磁矩最大,为负值,而轨道磁矩为正值,而第二层的Sm 原子的自旋磁矩和轨道磁矩都比第一层小,说明了Co 的3d 轨道与Sm 的4f 轨道发生了相互耦合,导致第二层Sm 的磁性降低,另个原因是计算轨道磁矩和各向异性必须要考虑相对论的效应。更有趣的发现,第一层Co 原子的平均自旋磁矩和平均轨道磁矩都比第二层的Co 原子自选磁矩和轨道磁矩低,更说明了轨道之间存在强相互作用和自选轨道的耦合作用。
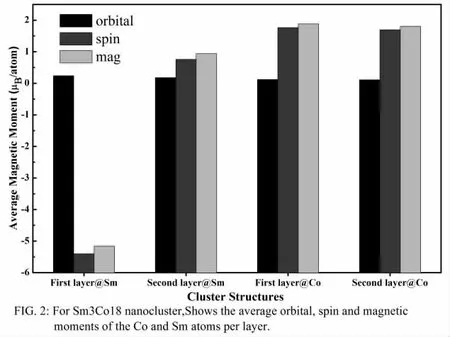
图2
综上所述,Sm3Co18 是核壳型双磁纳米颗粒能有效改善体系矫顽力和磁能积。在通常情况下,即使是性能优异的硬磁合金,其也要在室温以上的条件下工作,高温热激发将增强硬磁材料的磁化反转进程从而降低磁体的矫顽力和饱和磁化强度,进而大大降低硬磁体的磁能积[8],本文研究体系中Sm 原子与Co原子之间存在磁性反转,相当于引入具有高居里温度的软磁材料,并使其包裹在硬磁颗粒外围,充分结合硬磁相的高矫顽力及软磁相的高剩磁的优势。研究SmCo 纳米团簇的意义是纳米交换弹性磁体因其特有的硬磁- 软磁交换耦合而能同时拥有高的磁各向异性、饱和磁化强度、最大磁能积,最有潜力发展成为第三代高性能永磁材料,主要应用于电子电工、生物医学信息技术等领域的巨大潜力。
