硅胶整体柱的研磨与表面修饰制备新型高效液相色谱固定相
2020-05-30王照地王世革黄明贤
张 璐, 王照地, 刘 璐, 王世革, 黄明贤
(上海理工大学 理学院,上海 200093)
高效液相色谱在食品、医药、生命及环境等领域具有广泛的应用。硅胶基质以其机械强度高、粒径分布均匀、孔构造和比表面积易于控制、表面易改性、有机溶剂中不溶胀等优势,成为最常用的高效液相色谱填料[1-5]。近年来,硅胶整体柱由于其高通透性和快速分析的特点,在色谱研究领域受到日益广泛重视[6-15]。
在近期的研究中,硅胶整体柱主要是在毛细管柱内形成,由于柱子内径小,所能承受的柱流量很小,不适用于一般色谱仪器和色谱分析。但硅胶整体柱的制备方法已有大量的研究和报道,有很多可以借鉴的地方。将硅胶整体柱进行研磨和筛分,可以用来制备适合于当今常用的高效液相色谱分析的色谱柱填料[16-18],并且保存了微孔结构可调节和通透性高的优异特性。
本研究以聚乙二醇(PEG)为致孔剂,以四甲氧基硅烷(TMOS)为硅源,使其在醋酸和尿素的催化作用下水解缩合,从而形成硅胶整体柱;将此整体柱研磨后,再进行煅烧和浮选得到合适粒径的硅胶颗粒;最后通过表面修饰制备一系列新型色谱固定相。反相色谱是最常用的色谱模式,因此,本研究探索3 种新型反相色谱固定相的制备方法。另外,限进型色谱固定相在血液中的药物分析方面发挥着巨大作用[19-24],本研究也探讨了一种新型限进介质色谱固定相的制备方法。通过对所制备的新型色谱固定相进行色谱测试和应用评价,考察了基于硅胶整体柱的研磨与表面修饰方法制备高效液相色谱固定相的潜力。
1 实 验
1.1 材料与设备
四甲氧基硅烷(TMOS,纯度大于99%)、聚乙二醇(PEG,聚合度为10 000,分析纯)、硬脂酸乙烯酯(优级纯)、甲醇(HPLC 级)、四氢呋喃(THF,优级纯)和正十八硫醇(97%)购自上海阿拉丁生化科技股份有限公司。过氧化苯甲酰(BPO,优级纯)购自北京百灵威科技公司。尿素、冰醋酸、二氧六环、无水甲苯、咪唑、乙酸乙酯、乙腈、乙醇、偶氮二异丁腈(AIBN)、γ-(2,3-环氧丙氧)丙基三甲氧基硅烷(KH-560)、三乙胺、乙酸酐、四丁基氟化铵(以上均为优级纯)和乙腈(HPLC 级)购自国药集团化学试剂有限公司。7-辛烯基二甲基氯硅烷购自美国Gelest 公司。mPEG1000-SH(聚合度为1 000)和mPEG2000-SH(聚合度为2 000)购自上海梵硕生物科技公司。色谱应用测试样品如塑化剂、标准药物以及牛血清蛋白(BSA)等购自上海阿拉丁生化科技股份有限公司。
使用的仪器设备包括:230II 系列高效液相色谱仪和EC2006 色谱数据处理工作站(大连依利特分析仪器有限公司),150 mm×4.6 mm 色谱柱管和液相色谱装柱机(深圳正大流体机电设备有限公司);Tristar II 3020 比表面与孔径分析仪(美国Micromeritics 公司);VEGA3 扫描电子显微镜(SEM)(泰思肯贸易上海有限公司),SKL 箱式高温烧结炉(合肥晶科材料技术有限公司),KQ-300DE 型数控超声波清洗仪(昆山市超声仪器有限公司),电热鼓风干燥箱和真空干燥箱(上海—恒科学仪器有限公司),高速台式离心机(德国NUAIRE 公司),行星式球型研磨机(长沙科德仪器设备有限公司)。
1.2 实验方法
1.2.1 硅胶颗粒的合成
取4.86 g 聚乙二醇(PEG,聚合度为10 000)和4.95 g 尿素于聚四氟乙烯反应釜内衬中,用45 ml摩尔浓度为0.01 mol/l 的乙酸溶液溶解并进行磁力搅拌。10 min 后,加入5 ml 四甲氧基硅烷(TMOS),再继续搅拌40 min。将盛有混合透明溶液的内衬置于水热反应釜中,并在40 ℃的烘箱中温育48 h,之后再将烘箱调至120 ℃继续温育48 h。反应完毕降至室温后,得到白色固体产物。将此产物依次用水浸泡2 次,乙醇浸泡1 次,去掉液体后,将整块固体在80 ℃条件下干燥过夜,然后将此固体用研钵捣碎,再用球研磨机研磨(转速为150 r/min) 60 min,最后在550 ℃煅烧5 h。将煅烧后的硅胶颗粒在室温下分散于无水乙醇中,超声处理30 min,然后使悬浮液自然沉降,3 h 后去掉未完全沉降的上层,剩下的固体烘干保存。
1.2.2 含烯键C8 键合固定相
将煅烧和浮选好的硅胶颗粒置于70 ℃的鼓风干燥箱干燥48 h。取烘干后的硅胶颗粒5.61 g,通过干燥洁净的漏斗加入到250 ml 的三颈圆底烧瓶中,再加入150 ml 无水甲苯,接上搅拌器,调转速260 r/min,同时连接氮气,油浴升温至110 ℃搅拌并甲苯回流30 min 除水,之后加入3.6 g 咪唑。取3.6 ml 7-辛烯基二甲基氯硅烷加入上述混合溶液中,继续在通氮气环境下110 ℃回流并搅拌12 h。反应完毕后冷却至室温,过滤除去清液,用适量无水甲苯洗涤固体颗粒,再依次用甲醇、甲醇/水(50/50,体积比)混合溶剂、和甲醇离心洗涤。最后在鼓风干燥箱内70 ℃干燥6 h,得到含烯键的C8 键合固定相。
1.2.3 巯基-烯点击反应制备反相色谱固定相
取3.5 g 含烯键C8 键合的硅胶颗粒,通过干燥的漏斗加入到三颈圆底烧瓶底部,加入150 ml二氧六环,之后通氮气,温度调至70 ℃,加入1.36 g正十八硫醇和0.068 g 偶氮二异丁腈(AIBN),继续在70 ℃搅拌反应8 h。然后依次用二氧六环、甲醇离心洗涤2 次。所得产品置于70 ℃烘箱中干燥3 h,在60 ℃下真空干燥3 h。
1.2.4 硬脂酸乙烯酯表面聚合固定相
取适量碱性氧化铝分散在适量乙酸乙酯溶剂中,之后称量3 g 硬脂酸乙烯酯(Vinyl Stearate)溶于其中。将上述键合的含烯键C8 的硅胶放入250 ml三颈圆底烧瓶中,加入150 ml 乙酸乙酯,置烧瓶于油浴加热锅,搅拌速度为230 r/min,将上述硬脂酸乙烯酯单体溶液过滤到烧瓶中,随后用N2保护,加热油浴锅温度至65 ℃。待温度稳定,加入100 mg 引发剂BPO(过氧化苯甲酰),继续加热搅拌反应12 h。将得到的硬脂酸乙烯酯表面聚合固定相依次用乙酸乙酯和甲醇各洗2 遍。70 ℃烘箱中干燥3 h 后在60 ℃下真空干燥3 h。
1.2.5 限进介质色谱固定相的制备
取6 g 前期制备的硅胶颗粒加入到250 ml 三颈圆底烧瓶中,以200 ml 甲苯为溶剂,油浴锅加热至70 ℃并进行搅拌(速度为230 r/min),再加入10 ml的硅烷偶联剂γ-(2,3-环氧丙氧)丙基三甲氧基硅烷(KH560),70 ℃搅拌反应过夜。反应完成后,分别用甲苯、乙醇、乙醇/水和乙醇离心洗涤固体颗粒各1 次,得到带有环氧基的硅胶基质,鼓风干燥箱70 ℃烘干过夜备用。
取γ-(2,3-环氧丙氧)丙基三甲氧基硅烷键合的硅胶颗粒5 g,与200 ml 四氢呋喃一起加入到250 ml 三颈圆底烧瓶中,再加入2 g mPEG1000-SH 和1 g mPEG2000-SH,待固体全部溶解后,加入5 ml 的四丁基氟化铵溶液(1 mol/l THF 作溶剂),反应液在30 ℃搅拌3 h 后,将反应物用离心机离心分离,用乙醇洗涤3 次,在70 ℃恒温鼓风干燥箱中干燥6 h。
将上述反应产物分散于50 ml THF 中,加入3 ml乙酸酐和1 ml 三乙胺,在30 ℃继续搅拌反应3 h。分别用乙醇洗2 次,乙醇/水洗1 次,乙醇洗1 次离心洗涤,70 ℃鼓风干燥箱干燥9 h 得到一种限进介质色谱固定相。
1.2.6 装柱及测试
制备配比为1∶1 的异丙醇和三氯甲烷的混合液,以此为装柱匀浆液。取键合好的硅胶固定相2 g,加入上述匀浆液50 ml,超声2 min,然后在34.48 MPa 的压力下用甲醇通过装柱机将填料装入内径4.6 mm、长150 mm 的色谱柱中。
1.2.7 色谱测试
以尿嘧啶、苯乙酮、甲苯、乙苯、萘和芴等标准化合物为样品对装好的色谱柱进行反相色谱测试:流动相为甲醇和水;UV 检测波长为254 nm。用塑化剂标准物质或将标准药品分散在牛血清蛋白(BSA)中作为应用测试样品,色谱条件列于相应的色谱图说明中。
2 结果与讨论
2.1 硅胶基质颗粒的制备
硅胶整体柱的制备可以借鉴很多毛细管内硅胶整体柱的制备方法[6-15],其中,整体柱的制备条件决定了硅胶骨架的结构,从而决定了研磨后硅胶颗粒的大小和形貌。根据毛细管内硅胶整体柱制备方面的文献和前期的实验摸索,采用以聚乙二醇为致孔剂,四甲氧基硅烷为硅源,在醋酸和尿素的催化作用下水解缩合的方法来制备硅胶整体柱。将此整体柱研磨后,再进行煅烧和浮选,得到合适粒径的硅胶颗粒。研究发现,在控制反应时间在48 h、反应温度为先40 ℃然后升温至120 ℃、研磨速度为150 r/min、研磨时间为1 h 等条件下,可以得到如图1 所示的硅胶颗粒。从图1(a)中可以看出,硅胶颗粒保留了硅胶整体柱的骨架部分,粒径在4~6 μm 之间,并且从图1(b)中可以清楚地观察到硅胶颗粒的粗糙多孔表面结构。这种硅胶颗粒基本满足了色谱用硅胶基质的均匀粒径(5 μm 左右)和多孔性结构的要求。

图1 硅胶颗粒的扫描电子显微镜(SEM)图像Fig.1 Scanning electron microscope(SEM)images of silica particles
2.2 反相色谱固定相的制备
反相色谱是高效液相色谱中最常用的色谱模式,一般以C18 键合固定相为主,本研究试图制备3 种新型反相色谱固定相。首先,以甲苯为反应分散剂,咪唑为催化剂,通过7-辛烯基二甲基氯硅烷与硅胶的表面硅羟基之间的键合反应制备含双键的C8 键合固定相。咪唑是一种弱碱性化合物,可以中和反应过程中生成的盐酸。7-辛烯基二甲基氟硅烷(C8)通过稳定的Si—O—Si—C 键合到硅胶表面,如图2 所示。
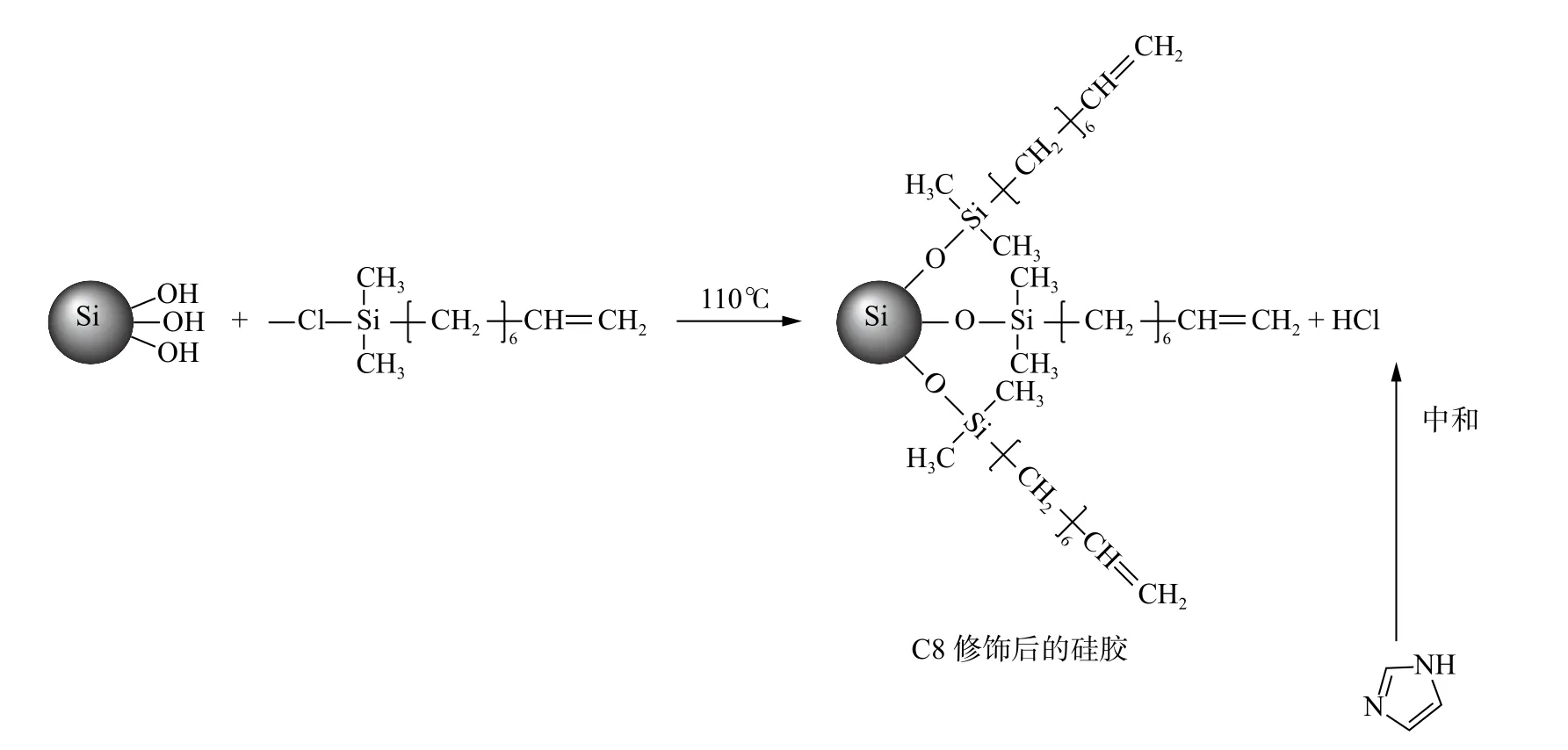
图2 含双键C8 固定相制备示意图Fig.2 Schematic diagram of the preparation of vinyl group containing C8 stationary phase
巯基-烯点击反应是常用的表面修饰和嫁接的方法[25-26],这种方法可以使材料表面改性,也可用来制备色谱固定相。本研究中通过硫醇-烯点击反应在上述制备的含双键的C8 键合固定相上进一步链接正十八硫醇,期望以此增加硅胶表面的疏水性,反应示意图如图3 所示。
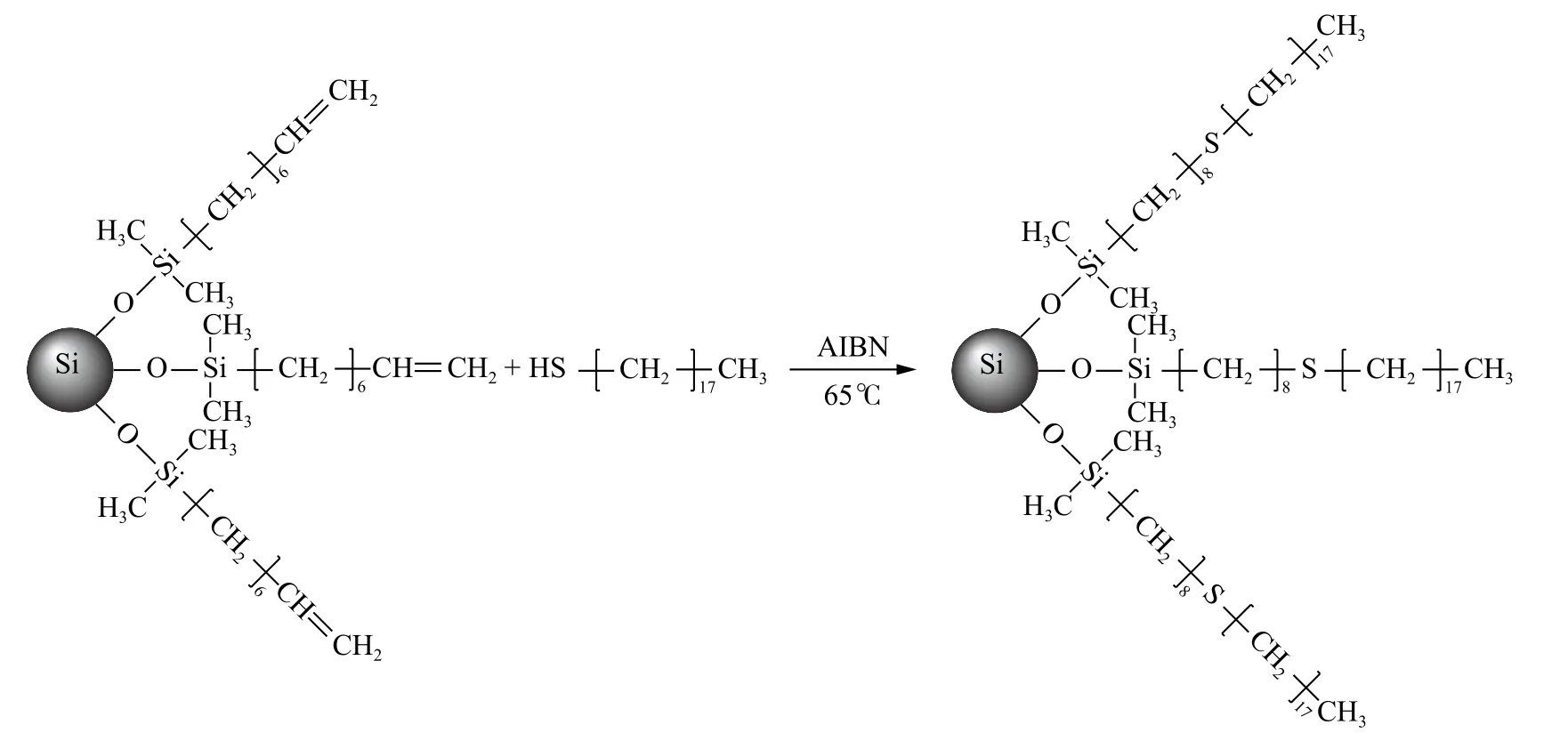
图3 通过硫醇-烯点击反应制备反相色谱固定相示意图Fig.3 Schematic diagram of the preparation of stationary phase via thiol-ene click reaction
另外,聚合物键合和涂覆的硅胶固定相也得到广泛的使用[27-28],这种表面修饰方法可以进一步增加硅胶固定相的选择性。本研究中将硬脂酸乙烯酯通过自由基共聚合反应接枝到前期制备的含双键C8 键合的硅胶表面,反应过程如图4所示。
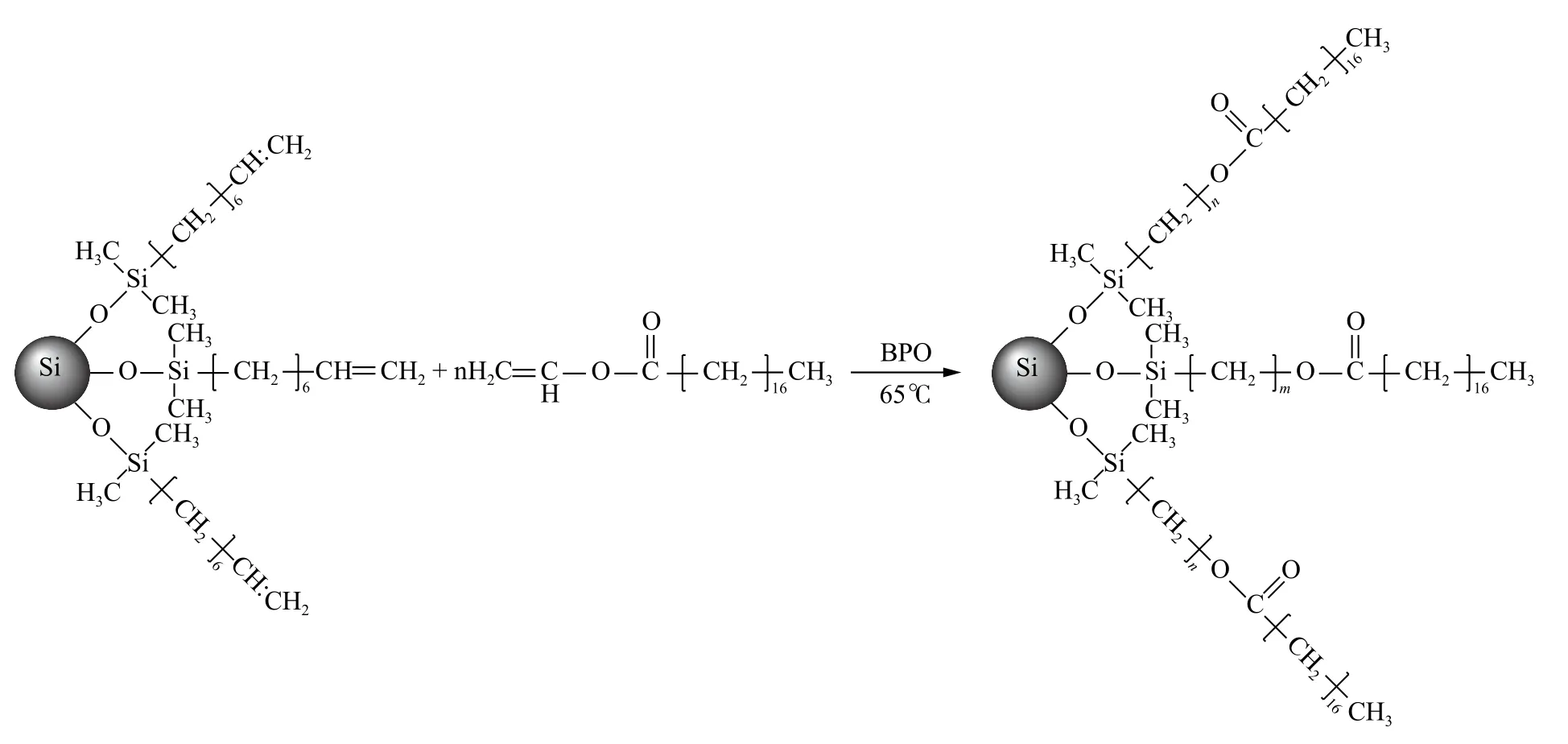
图4 硬脂酸乙烯酯聚合固定相制备流程示意图Fig.4 Schematic diagram of the preparation of vinyl stearate polymerization stationary phase
在图4 的反应中,过氧化苯甲酰作为自由基聚合引发剂,硬脂酸乙烯酯作为聚合反应单体,与硅胶表面键合的7-辛烯基中的双键发生自由共聚合反应,形成硬脂酸乙烯酯聚合物固定相。由于每一个表面双键都有可能嫁接上n(n>1)个硬脂酸乙烯酯单体,这种接枝聚合方法能进一步增加硅胶表面的疏水性能,从而增加其反相色谱保留和分离选择性。
2.3 反相色谱固定相的测试
将上述3 种色谱固定相的反相色谱行为进行测试和对比。在相同的洗脱条件下,相比于C8键合固定相(图5(a)),通过硫醇-烯点击反应链接的正十八硫醇固定相显示了增强的色谱保留(图5(b));而硬脂酸乙烯酯修饰的固定相对分析标准物质的保留时间更长且分离效果更好(图5(c))。这些色谱分离结果间接证明了硅胶表面发生了本文所设计的表面修饰化学反应。同时,这3 种色谱柱的柱前压都比一般球型5 μm 硅胶色谱柱的柱前压要小,证明了这种硅胶基质通透性好的优点。
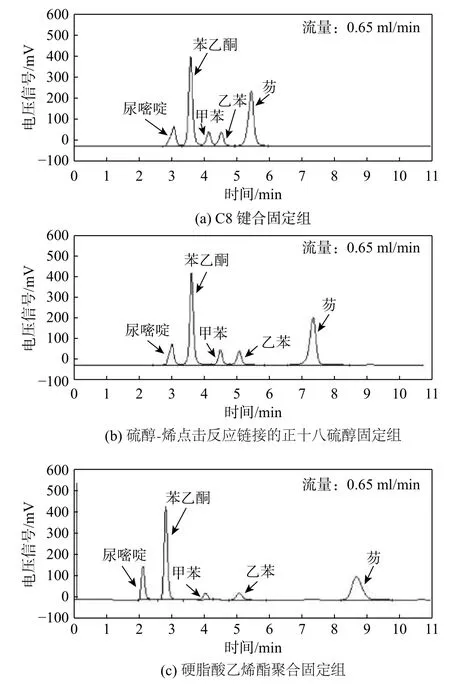
图5 反相色谱测试比较Fig. 5 Comparison of reversed phase chromatograms
考察了新制备的反相色谱固定相对标准塑化剂化合物的色谱分离效果,结果如图6 所示。在梯度洗脱条件下,可以看出3 种常见的塑化剂化合物都得到了非常高效的分离。塑化剂为一类对人体有害的化合物,在某些饮料和酒中曾发现了少量塑化剂。本研究所制备的色谱填料可以为塑化剂的色谱分析提供帮助。
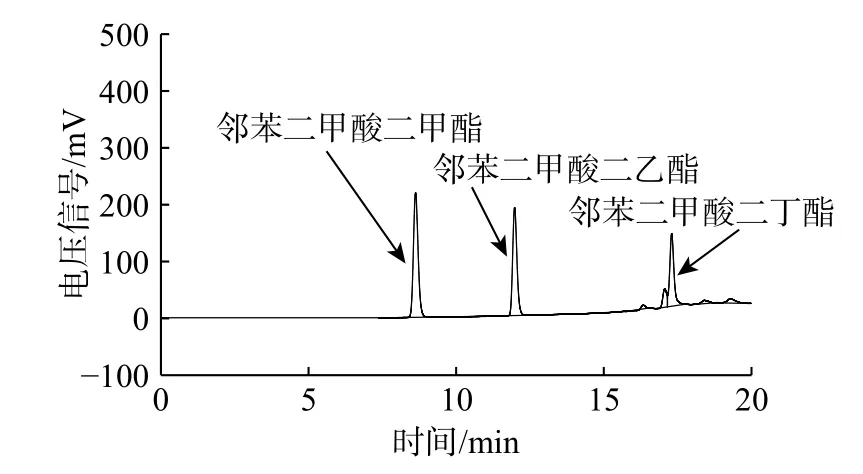
图6 反相固定相用于塑化剂的应用测试Fig. 6 Separation of common plasticizers with reverse stationary phase
在色谱基质的性能测试中,范德姆特(Van Deemter)曲线可以很好地反映出硅胶颗粒的特征及性能。图7 为测得的Van Deemter 曲线,从图中可以看出,随着流速的增加,乙苯的塔板高度先减小后增加,最后趋于平缓,在0.4 ml/min 时达到最小(塔板高度越小,塔板数越大,柱效越好)。这种硅胶材料由于其特殊的形状,在高流速状态下,塔板高度变化不大,并没有明显增加的趋势,因此,可以进行快速的HPLC(高效液相色谱)分析,体现了此种硅胶颗粒之高通透性的优势。
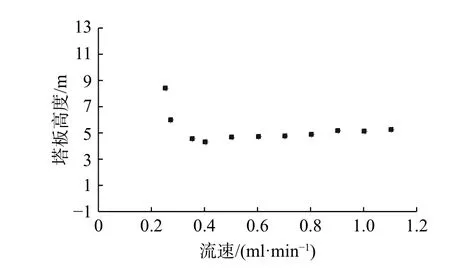
图7 乙苯在C8 反相色谱柱上的范德姆特曲线Fig.7 Van Deemter curve of ethylbenzene on C8 stationary phase
2.4 限进介质色谱固定相的制备和测试
将硅胶整体柱研磨、煅烧和浮选得到的硅胶颗粒,首先与KH560 发生键合反应,再与含巯基的PEG 分子通过巯基-环氧反应在硅胶表面链接PEG[29-30],最后,用乙酸酐与硅胶表面的羟基反应生成乙酸酯,从而得到一种新型限进介质色谱固定相。图8 是反应流程示意图。
这种限进介质色谱固定相的特点是最外层的长链亲水PEG 分子阻碍了蛋白质大分子与其内层疏水基团的作用,即达到蛋白质限进的作用,所以称为限进介质。但是,一般药物小分子可以渗透到内层,有一定的反相色谱保留。首先对新制备的限进介质固定相进行了反相测试,色谱测试条件中,流速为0.8 ml/min,流动相为甲醇/水(体积比为40/60),紫外检测波长为254 nm。其中,色谱峰鉴定分别有:a. 尿嘧啶,b. 乙苯,c. 萘。结果显示,这种色谱柱疏水性不强,只有当水在移动相的比例较高时,才有一定的色谱保留,结果如图9 所示。对于限进性固定相,移动相一般含水量大,所以,新制备的限进介质固定相符合相应的色谱分析要求。
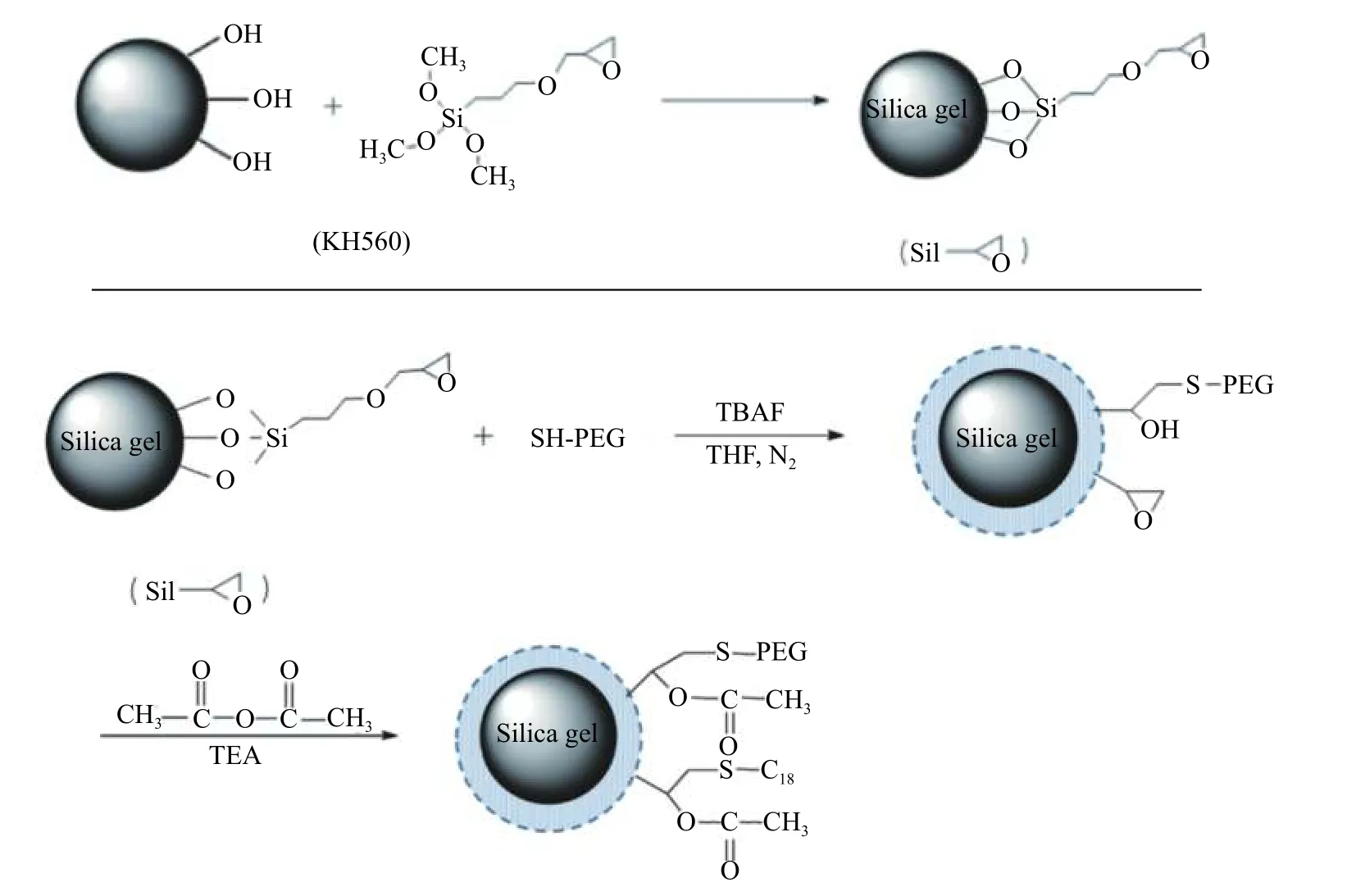
图8 限进介质固定相制备流程示意图Fig.8 Schematic diagram of the preparation of restricted access media stationary phase
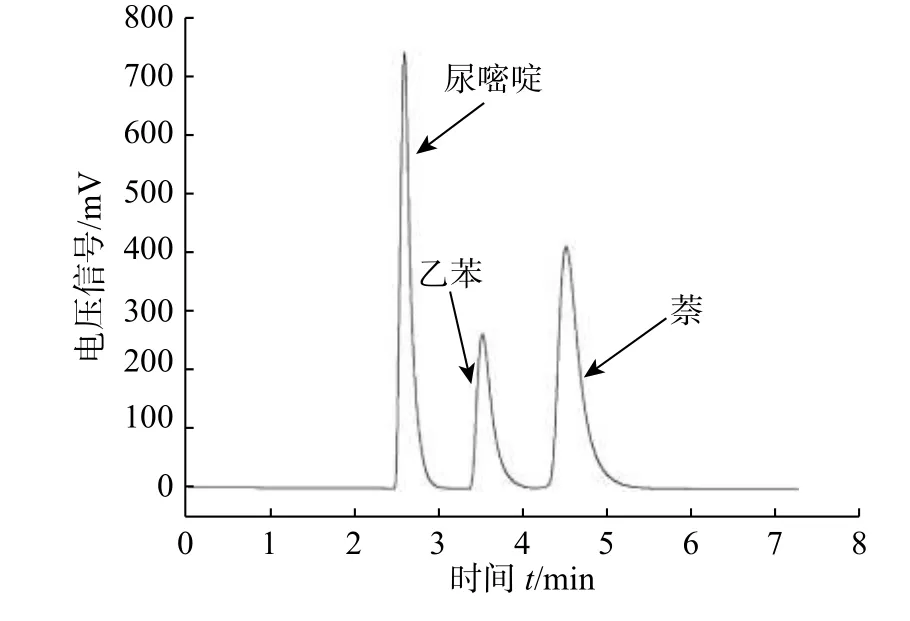
图9 限进介质固定相的反相色谱测试Fig. 9 Restricted access media stationary phase reverse phase test
限进介质色谱固定相在药物代谢分析中得到了广泛应用[19-24]。一般要求是将血液中的药物过滤后直接进入色谱柱进行色谱分析,血清中的大量蛋白一般以不保留的方式首先流出,被分析药物会渗透到固定相的内部,并在色谱柱上通过反相色谱作用进行分离。本研究所选的2 种药物分别为酮洛芬和依托度酸,其分子结构如图10 所示。
将上面2 种药物分散在牛血清蛋白BSA 中,直接进样分析,得到的色谱图如图11 所示。可以看出,在所制备的限进介质色谱固定相上,以流动相为PBS(pH 3.5)/ACN(80/20, 体积比),流速为0.8 ml/min,紫外波长220 nm 的检测条件下。BSA 无保留地流出,而2 种药物得到了较好的保留和分离,与前述的限进介质分离原理一致,可实现这些药物在血清中的定性定量分析。
图10 酮洛芬和依托度酸的分子结构Fig. 10 Strucutres of ketoprofen and etodolac
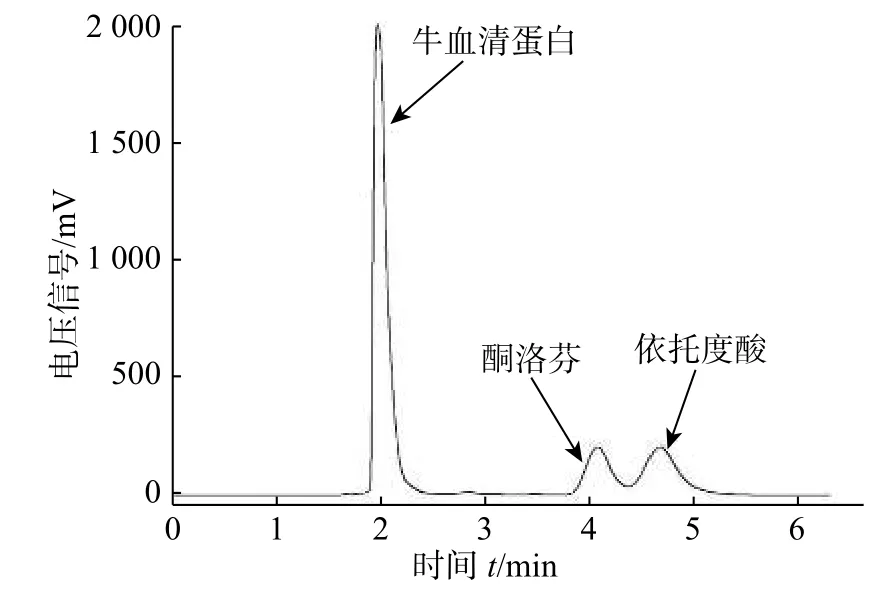
图11 限进介质固定相HPLC 分离BSA 中的酮洛芬和依托度酸Fig. 11 HPLC for the separation of ketoprofen and etodolac drugs in BSA on the restricted access media stationary phase.
作者还进一步测试了本研究所制备的硅胶键合前后的比表面积、孔体积和孔径的变化,结果列在表1 中。由表1 可以看出,键合前硅胶颗粒的孔径在16 nm 左右,键合后比表面积和孔径都有所减少,说明硅胶孔表面形成了一薄层键合物质。

表1 硅胶基质及KH-560 键合硅胶的BET 和BJH 测试数据Tab.1 BET and BJH measurement data of the as-prepared silica and KH-560 bonded silica
3 结 论
通过整体柱制备、研磨、煅烧和浮选,得到不定型的介孔硅胶颗粒,颗粒大小在4~6 μm,孔径约为16 nm,适合用作当今一般高压液相色谱的填料。采用本文的方法制备硅胶基质,方法简便、成本低、性能好。另外,通过不同表面修饰方法,可以利用本研究得到的硅胶制备出一系列高效液相色谱固定相。这些新型固定相都显示出较好的色谱分离效果,并且在塑化剂的分析和血样中药物的分析方面显示出一定的应用潜力。
可以预计,本研究制备的不定型的介孔硅胶基质,通过其他不同化学键合手段或涂覆法表面修饰后,可满足不同的色谱分离分析的要求。随着色谱技术在生物、医药及环境等领域的广泛应用,新型色谱固定相的研究将发挥巨大作用。
