钒配合物的生物效应、分子机制和药物发现
2020-05-29董雅琼张悦杨晓达
董雅琼,张悦 ,杨晓达 *
(1.北京大学医学部药学院化学生物学系暨天然药物和仿生药物国家重点实验室,北京 100191;2.北京大学国家中医药管理局中药配伍减毒重点研究室,北京 100191)
钒是重要的生命必需元素之一,在元素周期表中处于ⅤB族,属于过渡金属。钒的价层电子结构为3d34s2,具有+3、+4和+5多种化合价态,其中一些典型钒化合物结构如图1所示。在一些海洋生物中,钒可作为卤素氧化酶的活性中心,并以+3价的离子(V3+)存储于细胞中[1]。而在高等动物体内,钒通常以结合ATP的形式存在,但生理意义尚不明确。在细胞和生理介质中,钒主要以+4价的VO2+氧钒离子(vanadyl)配合物以及+5价的钒酸根(vanadate)或多聚钒酸根的形式存在。
根据离子的相似性作用原理[2],钒化合物具有广泛和多样的生物活性,可干预体内钙、铁和磷酸盐代谢过程和细胞氧化还原调节。因此,钒化合物具有多种药理活性,如降糖[3]、减肥[4]、心脏保护[5]、神经保护[6]和促伤口/骨愈合[7]等。目前对钒化合物类药物的研究主要集中于其抗糖尿病和抗肿瘤活性,特别是前者,得到了广泛的关注[8-9]。
1 钒化合物的抗糖尿病作用和分子机制
早在1899年,钒酸盐饮水就被用来治疗糖尿病患者[10]。20世纪80年代,钒化合物的类胰岛素样作用得到确认,因此成为抗糖尿病候选化合物而受到研究者广泛关注[11-12]。20世纪90年代,研究人员将联麦氧钒[bis(maltolato)oxovanadium,BMOV][13]及其乙基衍生物——乙基麦芽酚氧钒[bis(ethylmaltolato)oxovanadium,BEOV,研发代号:AKP-020]作为新型胰岛素增强剂(结构式如图1),开展了其临床治疗2型糖尿病的工作[3,14]。遗憾的是,AKP-020最终因商业和潜在安全性因素于2009年终止于Ⅱ期临床研究。
不过,在过去20年中,钒制剂作为食物补充剂以及2型糖尿病和并发症的替代药物而得到广泛应用[15]。多项体内外研究不断证实了钒化合物的降血糖作用[3]。钒化合物降糖制剂的优点包括:可口服、对1型和2型糖尿病均有效、降糖作用持久,且对防治糖尿病代谢综合征有益。实际上,钒化合物在降糖的同时,也发挥了胰岛β细胞保护[16-17]、心脏保护[5]、抗抑郁和神经保护[6]、改善脂代谢[18]、改善细胞抗氧化系统[19]和改善伤口愈合[7]等综合抗糖尿病疗效。
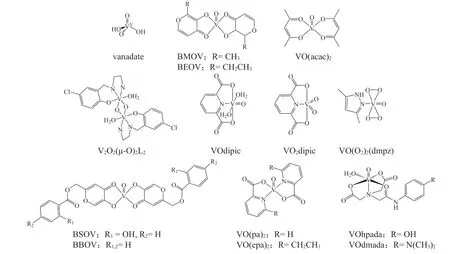
图1 典型抗糖尿病钒化合物的化学结构式Figure1 Structures of typical anti-diabetic vanadium compounds
在BEOV 的临床试验期间,其配合物的药动学(pharmacokinetics,PK)特性获得了很好的阐明[14]。总结目前的研究发现,钒化合物吸收后,根据其配合物的稳定性,会与生物介质发生配体交换。在细胞中,主要以大量的+4价离子及其配合物(90%~95%)及少量的+5价配合物(5%~10%)的形式存在[20-21]。氧钒配合物通常形成一水合配合物,几何结构在方锥体和八面体结构之间达到平衡[22-24]。在血清中,钒配合物及其水合形式可结合多种血清蛋白,如转铁蛋白、白蛋白和免疫球蛋白[25-26],主要以钒-配体-人血清白蛋白(VIVOL-HSA)三元配合物的形式存在[25]。白蛋白和转铁蛋白均能促进体内钒的转运,但白蛋白可能是更重要的药物载体[27]。钒配合物一般具有中等至良好的生物膜渗透性[表观渗透系数(Papp)> 10-6cm · s-1][28-29]。BMOV 的绝对口服生物利用度为21%~35%[24,30-31],与生物膜渗透性最好的双乙酰丙酮氧钒[bis(acetylacetonato)oxovanadium,VO(acac)2](结构式如图1)的生物利用度相当,这可能与钒配合物的药动学曲线上似乎存在的一个表观“存储池”有关。
钒化合物的有效降糖剂量介于0.05~0.6mmol ·kg-1· d-1,但当剂量超过0.2mmol · kg-1· d-1时,大多数钒化合物会导致食欲抑制,Malabu 等[32]发现,该作用抑制于下丘脑食欲中枢,而非胰岛素样作用,可能是钒化合物发挥减肥作用的原因。研究表明:钒化合物可在转录水平调节多种糖尿病致病基因[33]的表达,包括糖代谢、脂代谢、氧化应激、肌肉结构、蛋白质分解和生物合成等代谢功能基因组。钒化合物的作用主要表现为胰岛素增强和组织/细胞保护,其作用的主要分子途径包括促胰岛素信号转导、降低胰岛素抵抗、改善糖脂代谢(机制总结如图2所示)、胰岛素β细胞保护和神经保护作用。
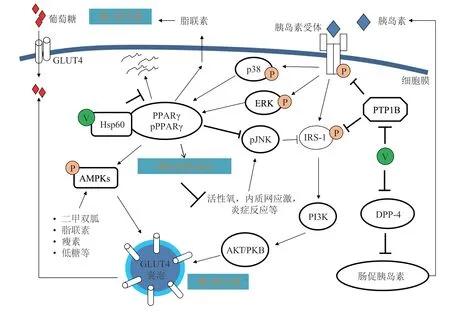
图2 钒化合物的胰岛素增强作用的机制Figure2 Insulin enhancement mechanism of vanadium compounds
1.1 促胰岛素信号转导
胰岛素由胰腺β 细胞分泌(见图2),到达目标组织后,胰岛素首先与胰岛素受体(insulin receptor,IR)结合,IR 是跨膜糖蛋白受体的α 亚单位,形成受体-配体复合物。然后激活β亚单位上的酪氨酸激酶并触发对接蛋白的激活,即胰岛素受体底物(insulin receptor substrate,IRS)[34]。随后,IRS激活磷脂酰肌醇3激酶(PI3K),从而导致蛋白激酶B(AKT/PKB)的激活,进而激活葡萄糖转运蛋白4(GLUT4)。此外,PI3K-AKT/PKB信号可能刺激重要的代谢功能,如脂类、蛋白质和糖原的合成。总的来说,胰岛素信号促进细胞中葡萄糖的利用。
钒化合物可能与胰岛素信号转导中的几个靶点相互作用,其中一个公认的靶点是蛋白酪氨酸磷酸酶1B(PTP1B),它是一种通过脱磷酸化作用对IR 和IRS的活性进行负调节的因子。研究表明:与野生型小鼠相比,持续高脂饮食的PTP1B基因敲除小鼠对肥胖的抵抗力和胰岛素敏感性增加[35],因此PTP1B被认为是治疗2型糖尿病的一个有希望的潜在治疗靶点。
钒酸盐是多种磷酸酶的非特异竞争性强抑制剂[36-37]。钒化合物抑制PTP1B的机制是与酶活性中心结合。其结合模式是钒酸根呈三角双锥构型,赤道氧原子与PTP1B的p环结合,顶部位置分别与半胱氨酸残基Cys215的硫原子和底物肽的酪氨酸氧相结合;研究发现,基本上钒化合物-磷酸酶复合物中,只有钒酸盐保留在特征结构中[37]。这表明,当钒配合物与磷酸酶作用时,无论其原始结构如何,配体的主要作用是将钒酸根传递到酶[36]。即使+4价钒配合物BMOV 等,在与PTP1B相互作用时,其与酶活性中心结合的氧钒离子实际上都发生了形态变化,转化为钒酸根。此外,大多数钒配合物具有与钒酸盐类似的抑制活性,其抑制常数(Ki)范围差别在1000 倍以内[36]。不过,钒配合物似乎在蛋白酪氨酸磷酸酶的同工酶[T 细胞蛋白酪氨酸磷酸酶(T-cell protein tyrosine phosphatase,TCPTP),巨核细胞蛋白酪氨酸磷酸酶2(megakaryocyte protein tyrosine phosphatase2,PTPMEG2),含C-Src 同源序列的酪氨酸磷酸酶SHP-1、SHP-2]中,对PTP1B的特异性略高[38]。只是由于总体亲和力水平还是不够高(lgKi为5~7),当钒化合物浓度在20μmol · L-1或以上时,会诱导活性氧(ROS)水平升高[39-44],导致活性中心的Cys氧化失活,从而使其作用复杂化。为此,Crans[37]观察到底物肽DADEYL 存在下,钒酸盐抑制剂的亲和力远高于所有其他钒化合物,据此提出通过利用有机配体和磷酸酶之间的额外相互作用,可以显著提高钒配合物的抑制效力和作用特异性。这一策略有望成为钒配合物磷酸酶抑制剂设计的通用策略。
PTP1B的抑制曾被认为是钒化合物胰岛素增强效应的主要原因。然而,PTP1B基因敲除大鼠的表型与用钒化合物[37]治疗的糖尿病大鼠表型并不相同。因此,抑制PTP1B只是钒化合物的作用机制之一,尤其是高浓度下的快速降糖作用。
1.2 降低胰岛素抵抗
胰岛素抵抗与肥胖和2型糖尿病密切相关,2型糖尿病最初可能表现为高胰岛素血症,但随后表现为胰岛素不足和胰岛素抵抗[45]。研究表明:胰岛素抵抗是炎症、内质网(endoplasmic reticulum,ER)应激、氧化应激、线粒体功能障碍和自噬失调等复杂相互作用的结果;其中,应激激活的c-Jun N 末端激酶(JNK)已越来越被认为是胰岛素抵抗的中枢介质[46-47]。JNK 是3种主要的丝裂原激活蛋白激酶(MAPK)之一,可能是应激与代谢性疾病之间的关键联系蛋白。JNK 被磷酸化激活后,导致IRS-1在一些丝氨酸残基如Ser307的磷酸化增加,从而抑制下游PI3K-AKT/PKB信号传导过程(见图2);抑制JNK 活性能改善胰岛素抵抗和葡萄糖耐受性[46-47]。笔者课题组发现,钒配合物在体内和体外均可抑制肝脏、脂肪和肌肉组织中的JNK 活化[48-50]。Niu 等[50]发现,VOdmada(结构见图1)可有效改善B6·BKS(D)-Leprdb/JNju (db/db)胰岛素抵抗小鼠的高胰岛素血症,同时提高其葡萄糖耐受性并使血糖水平正常化。钒配合物抑制JNK 的分子机制可能与调节未折叠蛋白反应(unfoldedproteinresponse,UPR)和过氧化物酶体增殖物激活受体γ(PPARγ)有关。
钒配合物也能抑制二肽基肽酶-4(DPP-4),分子模拟表明,钒配合物可以很好地嵌入DPP-4激酶域的活性位点裂缝中[51]。DPP-4是一种负责肠促胰岛素[胰高血糖素样肽-1(glucagon-likepeptide 1,GLP-1)和葡萄糖依赖性促胰岛素多肽(gastric inhibitory peptide,GIP)]降解的抗原酶;DPP-4抑制剂可延长体内肠促胰岛素效应,导致胰岛素分泌增加,胃排空减少,胰高血糖素降低,从而降低血糖水平[52-53]。DPP-4目前是降糖药(如西他列汀)和某些生物活性药物如黄连素的靶点。
1.3 改善糖脂代谢
研究表明:钒化合物可作为一般转录调节剂,并使多种糖尿病致病基因的蛋白表达正常化,其中包括许多葡萄糖/脂类代谢酶,如肝葡萄糖转运蛋白2、激素敏感脂肪酶、肌肉己糖激酶、磷酸烯醇式丙酮酸羧激酶、丙酮酸激酶[54-56]。钒化合物作为降糖药,不仅可以降低血液循环中葡萄糖水平,而且可以改善脂质代谢,例如降低血脂水平(即三酰甘油和胆固醇[3,54,57]),纠正低脂血症[57],降低游离脂肪酸(FFA)水平[4]。
钒化合物降血糖和降血脂双重作用的分子机制尚未完全阐明。然而,PPARγ 最近被认为是一个关键的作用靶点[48-49,58]。PPARγ 调节脂肪细胞分化、脂/葡萄糖代谢和炎症过程[59-60],可通过2种途径激活:胰岛素等激素[61-62]和PPARγ 配体激动剂[如噻唑烷二酮类(thiazolidinedione,TZD)[63-65]。
激活PPARγ 可改善2型糖尿病患者全身(脂肪、骨骼肌和肝脏)胰岛素敏感性[63-65]。不过,脂肪组织被认为是TZD介导胰岛素敏感性作用的主要靶组织。在脂肪组织中,PPARγ 活化促进脂肪生成,从而降低血液循环中FFA 的水平。在成熟脂肪细胞中,PPARγ 活化诱导许多参与胰岛素信号转导的基因的表达并增加了脂联素的产生。脂联素是一种众所周知的胰岛素增敏激素,可激活AMP激活蛋白激酶(AMPK)信号且同时减少引起胰岛素抵抗的TNF-α 和抵抗素的产生。此外,PPARγ 还可以降低许多组织,特别是心血管内皮的炎症反应。因此,PPARγ 在糖尿病和其他代谢性疾病(如血脂异常、脂肪变性等)的药物发现中一直是有吸引力的靶标。
钒化合物可在纳摩尔至微摩尔浓度调控PPARγ的表达和磷酸化修饰[50,66]。例如,在db/db小鼠中,其可上调脂肪、骨骼肌和胰腺组织[48,50,58]中的PPARγ 水平和下游基因如脂联素[49,58]的表达,进而在脂肪和骨骼肌中激活AMPK 和PPARα 信号转导、促进能量平衡和糖脂代谢[49,58](见图2)。然而,在肝组织[50]和3T3-L1前脂肪细胞[4]中,钒配合物则降低PPARγ 水平。其中的生物学意义有待进一步阐明,推测可能与抑制脂肪在肝脏沉积有关。但是在肝脏中,钒化合物仍然能增加AKT/PKB活化和抑制JNK 磷酸化[50],显示其降低了肝组织的胰岛素抵抗等活性。
钒化合物激活PPARγ 有2种可能途径。其一,氧钒离子能以高亲和力(lgKi=13~15)与相对分子质量为60000的热休克蛋白Hsp60结合,诱导PPARγ-Hsp60蛋白相互作用[48-49]。钒诱导的PPARγ-Hsp60 相互作用可以阻止TNF-α 诱导的PPARγ 降解,从而提高PPARγ 的蛋白质水平;并且,钒化合物可增强胰岛素或其他PPARγ 配体诱导的PPARγ 磷酸化,而不改变磷酸化模式[48]。其二,钒化合物可抑制Wnt3a[67],后者能抑制PPARγ 及其靶基因表达[68]。钒抑制Wnt3a 的原因尚不清楚。不过,Wnt3a 和UPR[69]之间相互关联,推测氧钒离子可能通过调节UPR 信号转导抑制Wnt3a(见图3)。由于钒化合物诱导PPARγ 的上调和活化作用与TZD药物明显不同,因此钒化合物不会产生TZD药物副作用[64,70],这是钒化合物较PPARγ 配体激动剂的一个优势。
1.4 胰岛β细胞保护
钒化合物对1型糖尿病[如链脲佐菌素(streptozocin,STZ)诱导的糖尿病]具有有效和长期的降血糖作用。研究发现,治疗后的动物都部分恢复了胰腺功能[71]。研究表明:钒化合物治疗维持STZ 诱导的糖尿病大鼠的胰岛β细胞结构[72],并促进了β细胞的增殖和再生[73]。钒化合物预防胰岛萎缩的效果优于胰岛素[16-17],而与葡萄糖毒性的缓解关系不大[17]。因此钒化合物具有β细胞保护的作用,这是与普通降糖药物相比的一个优点。
钒化合物对胰腺β 细胞的保护作用有2种途径(见图3):1)BMOV 和其他钒配合物及其转化产物均与胰岛淀粉样多肽(islet amyloid polypeptide,IAPP)表现出高亲和力,并抑制IAPP 的病理聚集[74];2)氧钒离子调节细胞UPR,这是β细胞保护的主要机制[75]。
UPR 是细胞响应ER 应激等损伤的一类防御途径[76]。ER 是蛋白质折叠和加工、脂质合成和钙储存的细胞器。在应激条件下,例如错误折叠蛋白超载或Ca2+耗竭,各种ER 受体从分子伴侣免疫球蛋白重链结合蛋白/相对分子质量为78000(BIP/Grp78)的葡萄糖调节蛋白上解离,启动下游的信号转导。在轻度或暂时性的ER 应激中,蛋白激酶RNA 样ER 激酶Perk 途径被激活,导致蛋白质翻译减少和细胞增殖停滞,从而防止了进一步的蛋白质超载;同时,肌醇酶1-X 盒连接蛋白1(IRE1-XBP1)和(或)转录因子6(ATF6)等信号激活,从而上调多种蛋白质伴侣(包括BIP/Grp78)的表达,用于结合、修复和清除错误折叠蛋白。如果ER 应激过于强烈或持续过长(如肥胖状态),延长的IRE1将激活JNK 信号通路,从而导致胰岛素耐受。此外,转录因子4(ATF4)被激活,导致CCAAT/增强子结合蛋白同源蛋白1(CHOP1)活化。CHOP1 会引起抗凋亡线粒体蛋白Bcl-2 的下调,导致线粒体途径的细胞凋亡(见图3)。研究发现,ER 应激在胰腺细胞变性和胰岛素抵抗发展中起着重要作用;通过血管紧张素Ⅱ受体阻滞剂替米沙坦[77]抑制ER 应激,可使STZ 诱导糖尿病的发生率降低一半以上。
笔者课题组发现,钒配合物VO(acac)2在纳摩尔至微摩尔浓度范围内本身不会影响UPR,但可以促进FFA 诱导的BIP/Grp78的表达,且降低应激下的促凋亡蛋白CHOP水平;同时,钒配合物恢复了FFA 干扰的细胞内Ca2+稳态[75],从而显著降低了FFA 诱导的β细胞损伤。此外,钒配合物促进FFA诱导的Perk 信号转导,也导致胰岛素合成减少。上述结果一方面有助于保持ER 稳态、减轻ER 应激状态下的β细胞负担,同时确保已合成胰岛素的折叠和分泌,并减少作为1型糖尿病“自身抗原”来源的错误折叠胰岛素[78]。另一方面,Perk 通路的增强可抑制Wnt 活性,从而上调PPARγ水平[69]。
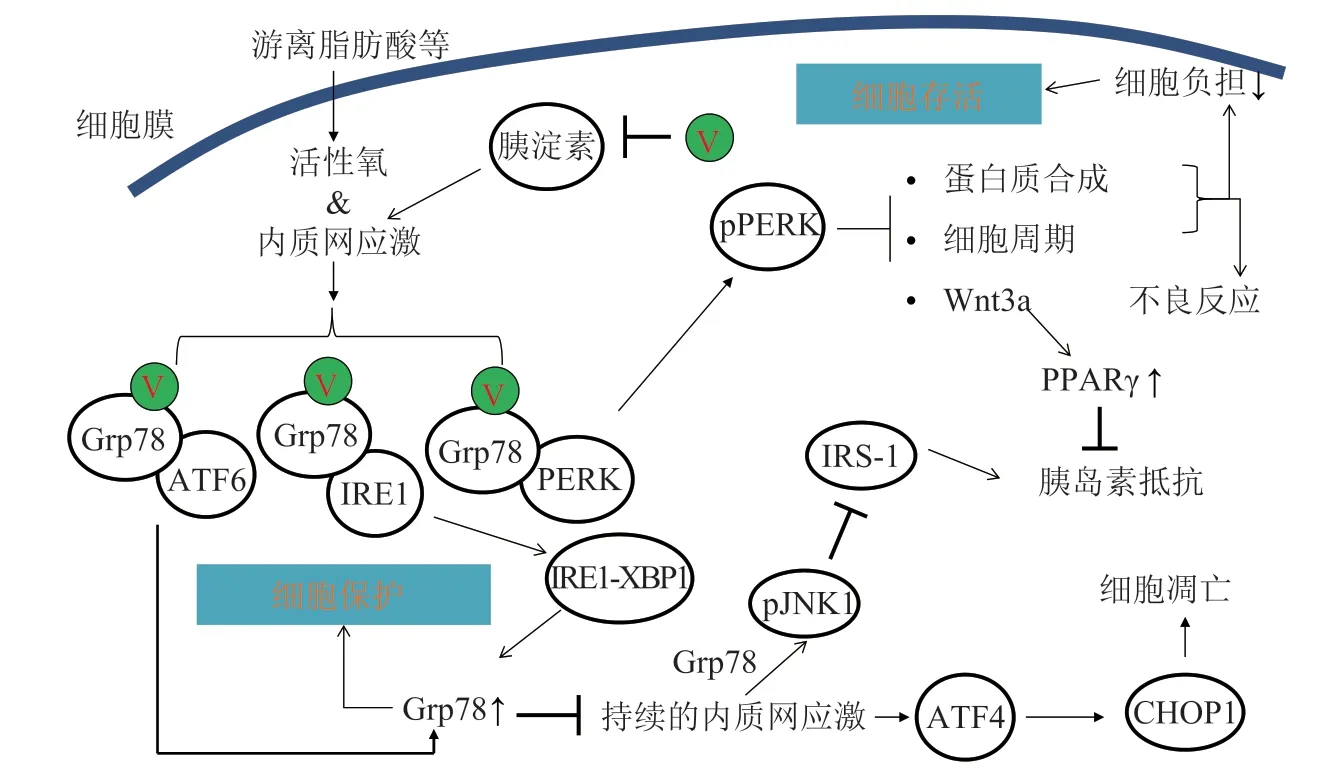
图3 钒化合物对未折叠蛋白响应的调控作用Figure 3 Regulation of vanadium compounds on the response of unfolded proteins
1.5 神经保护作用
阿尔茨海默病(Alzheimer disease,AD)已被证实与糖尿病密切相关[79-80]。2型糖尿病患者发生AD的风险比正常人群高约2倍。研究发现,AD和糖尿病存在许多共同的病理现象,相互促进彼此的病理进程[81-82]。超过80%的AD患者都存在明显的胰岛素抵抗,且血糖水平不正常。AD的重要病理因子β-淀粉样蛋白(amyloid β-protein,Aβ)可导致线粒体功能和动态损伤,激活JNK 和CDK5,从而抑制胰岛素信号转导;反之,胰岛素抵抗则导致细胞PI3K-AKT/PKB信号下降,导致糖原合成酶激酶GSK-3β 活性升高,从而引起Tau 过度磷酸化;而Tau 过度磷酸化诱导神经纤维缠结(NFT),这是导致神经元死亡的直接原因。此外,Aβ可与胰岛素竞争结合胰岛素降解酶(IDE),相互抑制对方的分解。因此,一些研究人员将AD称为3型糖尿病[80]。
笔者课题组发现,钒配合物可显著改善因高血糖而受损的空间学习和记忆[83]。研究发现,对APP/PS1 模型小鼠经口给予亚降糖剂量(0.1 mmol · kg-1· d-1)的VO(acac)2,明显改善了小鼠的认知表型,并减少了小鼠的神经元的缺失[84]。有趣的是,钒配合物的作用不受Aβ沉积的影响,也不减少APP/PS1小鼠脑组织Aβ斑块数量。不过,钒配合物显著减少了水溶性Aβ寡聚体,支持了水溶性Aβ寡聚体才是真正毒性Aβ物种的观点。机制研究表明(见图4):钒配合物是通过激活PPARγ-AMPK 的信号转导通路增强细胞糖代谢和能量生产,从而提高细胞活力,并进一步抑制GSK-3β活化、延缓进一步的Tau 病理发生。此外,钒配合物上调了线粒体伴侣蛋白Grp75的表达水平。Grp75增加不仅可抑制ROS产生,且可以抑制氧化应激诱导的p53核转位,进而抑制AD病理相关的神经细胞凋亡。抗糖尿病钒配合物的抗AD作用及其应用是未来值得深入研究的新课题。
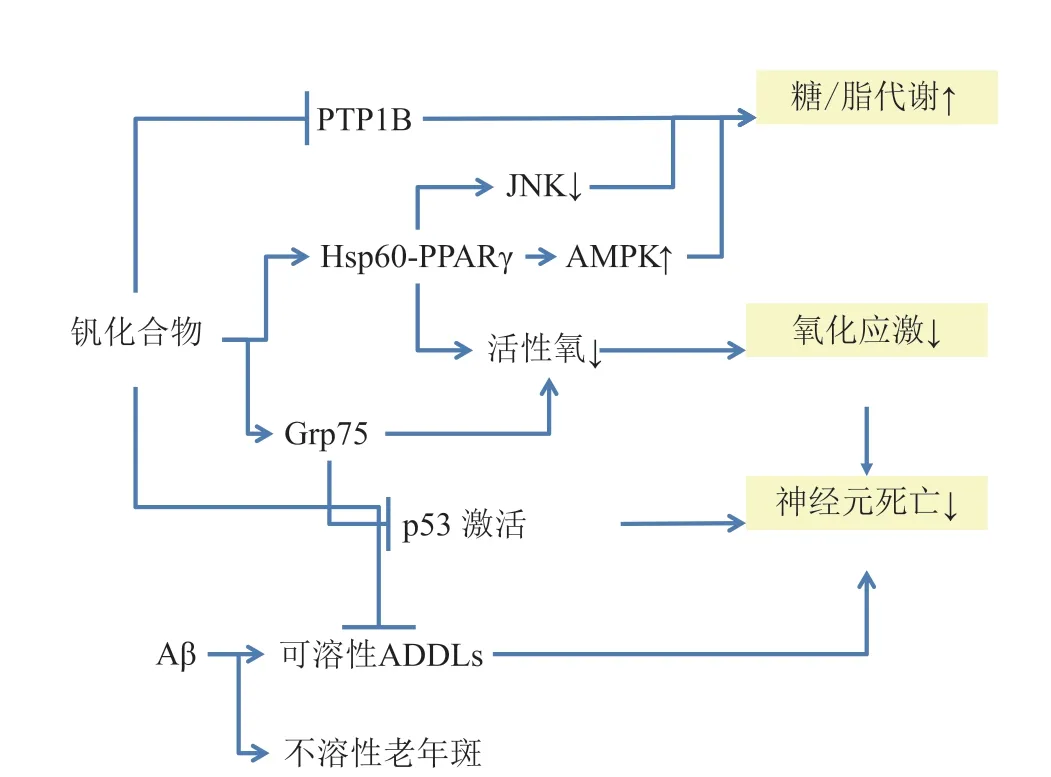
图4 钒化合物抗阿尔茨海默病作用机制Figure 4 Anti-Alzheimer’s disease mechanism of vanadium compounds
2 钒配合物的抗肿瘤作用及机制
很早发现,钒化合物具有显著的肿瘤预防作用,虽然这可能是由于上述调节机体能力代谢和抗炎、抗氧化等而发挥的作用,但研究亦发现钒化合物可减少DNA 的烷基化[85],从而降低DNA 突变的发生,这可能是钒化合物发挥肿瘤预防作用的机制之一。近10年来,钒化合物的抗肿瘤作用受到了研究者的关注。钒化合物能够抑制多种肿瘤的增殖和诱导肿瘤细胞凋亡,也能通过多种作用方式限制肿瘤细胞的侵袭和转移[86-87]。其具有多样化的抗肿瘤机制,主要包括:1)直接与DNA 骨架的磷酸基团结合,诱导DNA 损伤,但其结合方式不同于顺铂[88];2)促进活性氧(ROS)的生成,通过氧化应激相关信号途径导致细胞凋亡[88-90];3)通过增强c-fos相关信号转导或抑制某些关键磷酸酶,导致细胞周期阻滞[90-91];4)调节癌细胞能量代谢,抑制Warburg 效应[92];5)诱导癌细胞自噬[93];6)诱导肿瘤细胞失巢凋亡[90,94];7)通过与miRNA 结合,抑制肿瘤生长[95]等。
值得一提的是,钒化合物的抗肿瘤作用相比铂族药物来说一般作用较弱,但钒化合物诱导某些肿瘤细胞凋亡的能力明显大大高于正常细胞,并且对两者的作用模式也不同[42]。例如对HepG2肝癌细胞,VO(acac)2可通过诱导持续的ERK 磷酸化激活而降低磷酸化Rb蛋白水平,从而使HepG2细胞生长阻滞于G1/S期,进而诱导肿瘤细胞凋亡;而相同浓度的VO(acac)2并不影响正常L-02肝细胞的增殖。高浓度的VO(acac)2在正常肝细胞中通过诱导氧化应激导致细胞毒性,抗氧化剂N-乙酰半胱氨酸(NAC)处理可抑制这种毒性;但NAC处理不能抑制VO(acac)2对肿瘤细胞的毒性[42,96]。笔者课题组推测细胞周期调节机制的不同可能与抑制某些关键磷酸酶、导致蛋白质磷酸化修饰的差异有关[97]。细胞的生长调节因细胞类型和生长周期的不同而不同,因此增殖(肿瘤)细胞和非增殖(组织)细胞对钒配合物处理的反应存在差异。再如,钒配合物可对2 种细胞类型的干扰素的应答分别进行调节[98],从而使肿瘤细胞(而非正常组织)对干扰素和溶瘤病毒感染更为敏感[99]。因此,有望建立基于钒配合物预处理的新型抗癌生物疗法。
在各种肿瘤细胞中,神经母细胞瘤细胞系可能对钒化合物治疗最为敏感。最近笔者课题组发现,钒配合物对神经母细胞瘤细胞系的毒性(IC50为5~30μmol · L-1)与顺铂相当(IC50为0.6~40μmol · L-1),是对正常原代神经元细胞毒性(IC50约为250 μmol·L-1)的15倍。进一步的对比研究表明[97],钒配合物通过浓度依赖的2条途径抑制神经母细胞瘤生长:一是诱导细胞周期蛋白cyclin D过表达,通过cyclin D介导的未知途径诱导细胞凋亡;二是在高浓度时(>50μmol·L-1)抑制多种细胞周期相关的磷酸酶(钙调神经磷酸酶和Cdc25C)的活性,导致细胞周期阻滞和最后的细胞凋亡。这2条途径均为非p53依赖的,因此,钒配合物的凋亡诱导模式对发现新的抗癌药物、克服因p53突变引起的化疗耐药性很有意义。
值得一提的是,神经母细胞瘤是继白血病和脑癌之后儿童中第三常见的癌症[100]。在儿童癌症死亡病例中,约15%与神经母细胞瘤有关[101]。目前该癌症使用顺铂药物(顺铂、卡铂)与其他药物(如阿霉素)联合化疗。然而,顺铂药物对神经母细胞瘤细胞和正常神经细胞表现出相当接近的细胞毒性[96]。事实上,周围神经毒性一直是顺铂药物[102]的主要剂量限制性副作用[103-104]。因此,钒化合物在神经母细胞瘤等中枢神经系统肿瘤的治疗中具有较大的潜力。
3 钒配合物药物设计和药物发现
对钒化合物的生物学效应和分子机制的研究表明,钒化合物是继顺铂之后具有巨大开发潜力的金属药物。显然,钒化合物在动物体内外试验中均表现出有效的降血糖作用,并在治疗代谢综合征方面取得了成功,使其成为有前途的抗糖尿病药物。虽然人们对钒金属毒性的忧虑一时难以消除,但以往的研究表明,钒的毒性虽然复杂,但主要与金属引起的氧化应激有关[105],因此是可预计和可控制的。
笔者课题组对抗糖尿病钒化合物的理性结构设计曾有较全面的综述[106]。未来的重点依然是如何控制金属离子的体内物种变化。物种变化问题对于所有金属药物都是一个重要的问题和挑战[107]。在生物体内,过渡金属配合物都会发生复杂的化学反应和物种变化,例如水化、水解、配体交换、氧化还原反应和与蛋白质分子的非共价结合(之后常发生与蛋白质分子的配体交换)等。这些变化将导致药物失效或产生毒性,并且会导致配合物的体内活性与体外筛选结果严重不一致。
对于抗糖尿病钒配合物,如前所述,配体的作用是将活性氧钒离子或钒酸根传递到其靶酶/蛋白质[108-109],其中重要的靶蛋白为热休克蛋白家族(如Hsp60、Grp78和Grp75等)和蛋白磷酸酶PTP1B。钒酸根及目前已知的氧钒配合物与PTP1B的结合作用(lgKi为6~8)[36],与其对生物介质中各种分子的非特异性结合作用(通常为lgKi为4~7)相当。因此,如何设计与PTP1B更稳定和更特异性结合的钒配合物是未来设计抗糖尿病钒化合物的一个重要工作。而氧钒离子具有与Hsp60结合的高亲和性(lgKi为13~15)[106],从热休克蛋白家族的结构相似性推测,钒化合物与其他热休克蛋白家族靶蛋白的结合也应在此范围,因此,稳定常数(lgKs)在8~12范围内的氧钒配合物既可以避免非特异性结合,又能高效地向靶蛋白递送氧钒离子。总结以往研究的经验,笔者课题组提出钒配合物理性设计的要点为:1)氧化数为+4或+5的钒配合物,尤其是+4价的钒配合物,更适合于制备安全的抗糖尿病药物;2)配合物的稳定性高(lgKs为7~13),这样可以有效和特异地释放钒离子;3)配合物与白蛋白结合并保持结构完整,将使钒配合物在体内的分布具有良好的特性;4)具有抗炎和抗氧化活性的结构的引入,将有利于平衡治疗性能和副作用。笔者课题组最近制备的氨基三乙酸衍生物氧钒配合物VOhpada[58]和VOdmada 系列化合物,其lgKi约为11。与其他钒配合物相比,这些化合物的治疗指数均显著提高。
钒化合物的另一个潜在应用是抗肿瘤作用。抗肿瘤钒化合物的设计与抗糖尿病钒配合物的设计在许多方面相似,比如对特定磷酸酶的抑制剂设计,如何提高抑制剂与靶酶的亲和力及结合的特异性是一个巨大的挑战。特别是,目前机制研究虽然阐明了其作用的分子途径,但尚未明确其中的靶分子。因此,通过理性设计抗肿瘤钒配合物目前还存在巨大的困难,期待今后在钒配合物抗肿瘤药理分子机制领域的重大发现。
从目前已知的钒化合物药理作用的分子机制来看,钒化合物还有许多潜在的应用空间有待深入探索。例如抗糖尿病钒配合物的抗AD应用,进一步探索设计新型抗AD钒配合物。近来,笔者课题组意识到,从金属离子的相似性作用规律出发[2],探索金属离子调节体内应激响应是未来发现高效低毒金属药物的重要策略[105,110]。目前的研究表明,钒配合物与热休克蛋白家族具有较特异的相互作用,针对热休克蛋白家族研究钒化合物的生物活性及药理作用是未来钒配合物药物研究大有可为的领域。
4 结语
过去20年,研究者对钒化合物生物活性及其分子机制获得了较为深入的认识,推动了钒化合物特别是抗糖尿病钒配合物的药理机制研究和理性分子设计。钒配合物具有良好的药动学特性,在生理介质中,可以通过复杂的物种变化与各种细胞内靶标进行相互作用。研究表明,钒化合物可以通过改善葡萄糖和脂质代谢,促进胰岛素信号转导,发挥降血糖以及胰岛素保护作用。除了抗糖尿病药物,钒配合物在抗肿瘤和抗AD药物上也具有巨大的开发潜力。相关研究也将是具有巨大发展前景的研究领域。
