新型喹唑啉类化合物的合成研究
2020-05-25孟令强杨绍娟张小博
孟令强, 杨绍娟, 张小博
(1.燕京理工学院 教务处,河北 三河065201; 2.燕京理工学院 生命与健康学院,河北 三河065201;3.燕京理工学院 工学院,河北 三河065201)
靶向治疗是目前肿瘤治疗领域中的研究热点之一,大量研究证明,以EGFR(表皮生长因子受体)作为药物作用的靶点是靶向治疗的一个新思路[1-2].这个思路得到国际上许多研究机构和制药集团的重视,以EGFR作为作用靶点的药物研究已成为抗肿瘤药物研发的重要途径和方法.
迄今为止,关于EGFR 酪氨酸激酶抑制剂的研究很多[3-6],总结起来包括以下几类:黄酮和异黄酮类、肉桂酰胺类、苯乙烯类、亚苄丙二氰类、苯胺类和苯胺基-嘧啶类.过去通常的观点是各种受体的激酶区氨基酸序列是高度保守的,因此前几类EGFR酪氨酸激酶抑制剂很少具有特异性.
目前人们认识到受体激酶区的结构存在着微小差异,因此,通过对激酶ATP 结合区的研究已成功设计出几类能与EGFR 激酶区特异性结合的酪氨酸激酶抑制剂.其中,结构中含有4 -胺基喹唑啉、4 -芳基酰胺基吡啶并嘧啶和4 -苯基胺基吡咯嘧啶等的化合物受到化学家的广泛关注.在所有这些抑制剂中,4 -胺基-喹唑啉类化合物一直是人们最关注的,相关报道证明4 -胺基喹唑啉类化合物通过与ATP的结合位点结合,从而可以高度选择性地抑制EGFR的磷酸化[7-8].
根据EGFR抑制剂喹唑啉类化合物的结构特征,可以把该类化合物拆分为Ⅰ、Ⅱ和Ⅲ3 个片段.如图1 所示.
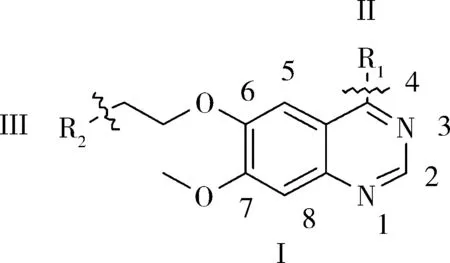
图1 喹唑啉类化合物的构效关系Fig. 1 Structure-activity relationship of quinazoline compounds
迄今为止,对高活性EGFR 抑制剂喹唑啉类化合物的构效关系的研究已取得丰硕的成果[9-11]:1)喹唑啉环的N -1,N -3 分别以氢键与ATP 结合,是抗癌的活性必须的结构(片段Ⅰ);若N -3被C取代,抑制力下降100 倍,N -1 被取代,抑制力则下降3 700 倍;2)喹唑啉的2 -位和8 -位空间位阻大,不宜引入基团,5 -位引入基团也不利于提高活性;3)4 -位引入基团(即片段Ⅱ)可提高活性;4)喹唑啉环中6,7 -位引入电子基团会提高整个化合物的抑制力.
笔者在前期研究中,制备出了新型喹唑啉类化合物的中间体2 -氨基-4 -甲氧基-3 -(3 -氯丙氧基)苯甲酸甲酯,以其为原料,经过环合、氯代和取代等反应,采用通过活性基团拼接法,制备出6种文献未见报道的新型喹唑啉类化合物,将为寻求活性更强的抗肿瘤药物打下基础.该法制备喹唑啉类产物可修饰位点多,具有操作简便、反应条件温和等优点.在药物化学中应用广泛,此类化合物保留了传统喹唑啉化合物的所有反应位点,且在4 -位和6 -位处引入了一个可修饰性较强的氯,将大大扩充产物的骨架丰富性,有助于发现新的具有良好生物活性的喹唑啉类化合物.
基于以上探索,本文设计并合成了6 种无文献报道的新型喹唑啉类化合物,合成路线如图2所示.

图2 喹唑啉类化合物的合成路线Fig. 2 Synthetic route of quinazoline compounds
1 结果与讨论
1.1 中间体1 的合成 本文首先尝试了文献[12 -13]采用甲脒盐酸盐进行环合的方法,但是副产物多;本文又尝试了文献[14]的方法,与甲酸进行环合反应,结果显示产物非常少,因为酰胺基较酯基更活泼;本文按照文献[15]方法采用醋酸甲脒,脒中的一个氨基进攻2 -氨基-3 -(3 -氯丙氧基)-4 -甲氧基苯甲酸甲酯中的酯基形成酰胺,另外一个氨基则与2 -氨基- 3 -(3 -氯丙氧基)-4 -甲氧基苯甲酸甲酯中的氨基发生胺交换反应成环.结果显示此方法操作简便,反应时间短,收率较高,达到91.586%.
1.2 化合物2 的合成条件 4 -(3 -氯-4 -氟苯胺基)-7 -甲氧基-6 -(3 -氯丙氧基)喹唑啉是合成喹唑啉类化合物的关键中间体,其反应是喹唑啉-4 -酮首先发生互变异构,由酮式(1)变为烯醇式(1′),再发生氯化反应.
本文按照文献[15]的方法,采用氯化亚砜回流,DMF 做催化剂反应,收率仅为61%,课题组又尝试以草酰氯、三氯氧磷和氯化亚砜作为氯化试剂,添加三乙胺、DMF 和吡啶做催化剂.实验发现:单独使用草酰氯、三氯氧磷和氯化亚砜为氯化试剂时反应时间长,收率为30%左右;加入三乙胺或DMF时收率提高至40% ~60%;采用氯化亚砜为氯化试剂,吡啶做催化剂反应副产物少,收率为70%以上,和其他氯化试剂相比有比较显著的优势(如表1 所示).

表1 氯代条件的优化Tab. 1 Optimization of chlorinated reaction
温度升高对氯化反应具有促进作用,反应温度较低时,收率相对较低;当反应温度升高至50 ℃以上时,反应时间明显缩短;当加热温度超过79 ℃(即氯化亚砜的沸点)时,收率呈下降趋势.原因可能是温度过高导致氯化亚砜的分解和副产物生成.由表1 可以看出,反应温度为79 ℃,反应1 h,收率最高,可达77.6%,在该温度下继续延长反应时间,收率无提高.
1.3 化合物3b 的合成条件 化合物2 与苯胺或硫醚反应不同,本文尝试了不同的溶剂以及后处理方法.以与3 -氯苯胺反应为例,本文通过实验得出以异丙醇为溶剂,以叔丁醇为重结晶溶剂时,收率和纯度最高(结果见表2).

表2 溶剂及反应时间的优化(回流反应)Tab. 2 Optimization of solvent and reaction time(reflux reaction)
1.4 化合物4a(4b,4c,4d)的合成条件 化合物3的6 -位侧链氯相对不活泼,该步反应参考袁立等[15]的方法,先用活泼原子取代氯,再由其他基团取代活泼原子,本文尝试了KI,收率较高,后处理方便简单.
1.5 化合物4e(4f)的反应条件 由硫醚转换成亚砜,本文采用活性二氧化锰和N -氯代丁二酰亚胺反应,收率为30%左右,增加氧化剂用量并延长反应时间,收率提高不明显;采用双氧水反应30 min,收率为50%左右,但增加双氧水用量或延长反应时间,收率反而下降,经结构鉴定发现部分产物由亚砜转化为砜;而采用过硼酸钠做氧化剂时,收率为90%左右.经实验得出:当过硼酸钠和硫醚的物质的量比为1∶ 1,反应时间为0.5 h 效果最佳(详见表3).
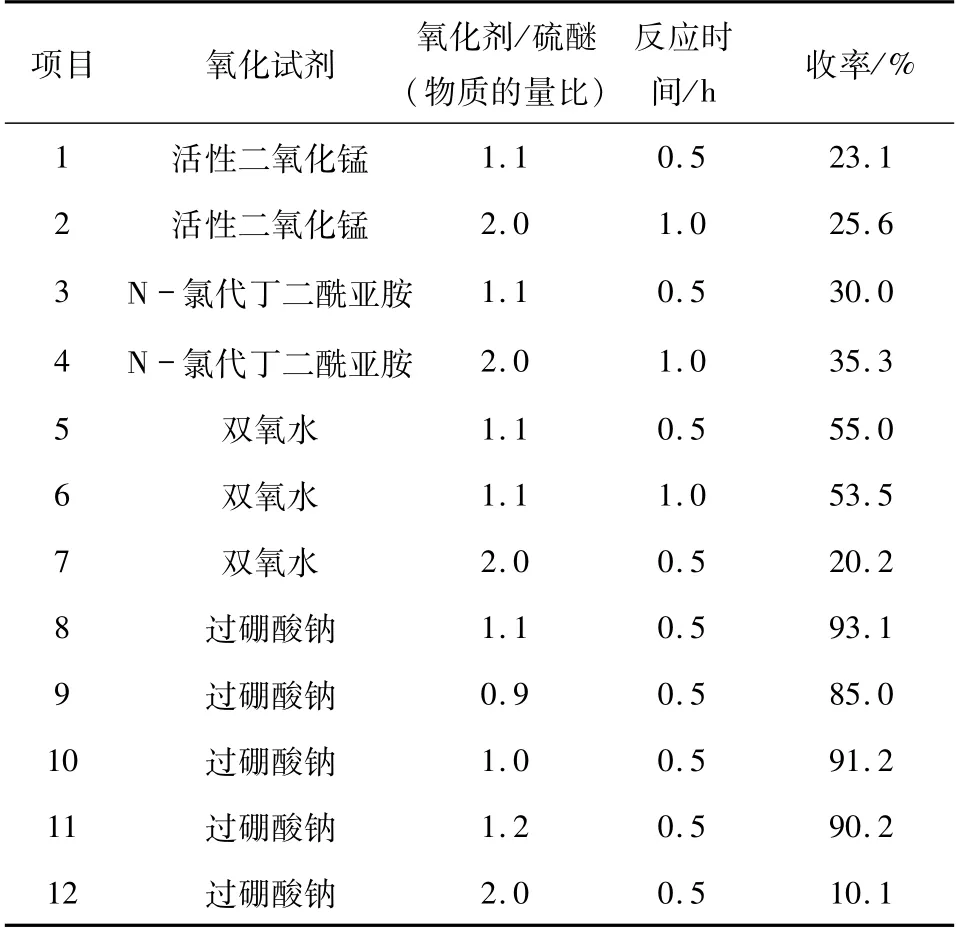
表3 将硫醚氧化成亚砜氧化条件的优化Tab. 3 Optimization of oxidation reagent
2 结论
本文设计并合成了6 个新型喹唑啉类化合物,均未见文献报道,并通过GC -MS 和1HNMR 确认了各化合物的结构.本文对各步反应条件进行了优化,最终得到一条反应条件温和,后处理方便,收率高,适合于工业化生产的工艺路线.
4 -位取代喹唑啉有着优异的活性,但是对于4 -位取代喹唑啉的研究,人们大多关注的是芳胺的取代,本文对此进行了创新,将新结构硫醚基团引入喹唑啉4 -位上,并将4 -位硫醚基团转变为亚砜,该反应无专利及文献报道,因此本文将硫醚及亚砜引入4 -位,以考察硫醚基团转变为亚砜对抗肿瘤活性的影响.
6 -位可以用不同的基团来取代,在课题组的前期研究中,发现片段1 -甲基-5 -三氟甲基苯并咪唑基-2 -硫醚基具有良好的抗肿瘤活性,因此本文将此片段引入到了新化合物中,以期获得更好的抗肿瘤物质.
3 实验部分
3.1 仪器与试剂 X -4 型数字显微熔点测定仪(温度计未经校正,北京泰克仪器有限公司);核磁共振仪(AV300,德国布鲁克公司),TMS 作为内标,氘代二甲基亚砜作为溶剂;MS 采用Shimanzu(QP2010 型)气相色谱-质谱联用仪测定;所用试剂均为分析纯的试剂.
3.2 实验方法
3.2.1 中间体的合成
1)7 -甲氧基-6 -(3 -氯丙氧基)喹唑啉-4 -(3H)-酮(1)的合成.
向100 mL 茄形瓶中,依次加入50 mL 无水乙醇、2 -氨基-4 -甲氧基-3 -(3 -氯丙氧基)苯甲酸甲酯(10.0 g,37.0 mmol)和醋酸脒(9.2 g,90.0 mmol),加完后回流反应2 h,TLC监测反应完全,冷却,抽滤,滤饼用乙醇重结晶,干燥得淡黄色固体9.0 g,收率91. 586%.熔点80 ~81℃.1H NMR
(CDCl3,300 MHz)δ(ppm):1. 85 ~2. 01(m,2H,CH2);3. 67(t,J =4. 0 Hz,2H,CH2Cl);3. 84(s,3H,CH3);4.05(t,J =6.0 Hz2H,CH2O);7.13(s,1H,ArH);7. 47(s,1H,ArH);9. 18(s,1H,ArH);12.26(s,1H,NH).
2)4 -氯-7 -甲氧基-6 -(3 -氯丙氧基)喹唑啉(2)的合成.
向100 mL茄形瓶中,加入30 mL氯化亚砜,室温搅拌下慢慢加入7 -甲氧基-6 -(3 -氯丙氧基)喹唑啉-4 -(3H)-酮1(10.0 g,37.0 mmol),再加入3 mL吡啶,加完后回流反应1 h,TLC 监测反应完毕,旋干反应液得到油状液体,再倒入50 mL冰水中,静置,有淡黄色固体析出,抽滤,水洗滤饼至中性,干燥得到类白色固体8. 293 g,收率77.6%.熔 点88 ~90 ℃.1H NMR(CDCl3,300 MHz)δ(ppm):1.85 ~2.01(m,2H,CH2);3.67(t,J =4.0 Hz,2H,CH2Cl);3.84(s,3H,CH3);4. 05(t,J =6.0 Hz,2H,CH2O);7.47(s,1H,ArH);7.66(s,1H,ArH);9.59(s,1H,ArH).
3)4 -(对溴苯硫醚基)-7 -甲氧基-6 -(3-氯丙氧基)喹唑啉(3a)的合成.
向100 mL茄形瓶中,加入50 mL异丙醇、4 -氯-7-甲氧基-6 -(3 -氯丙氧基)喹唑啉(10.0 g,35 mmol)和对溴苯硫酚(7.8 g,415 mmol),加完后回流反应30 min,TLC监测反应完毕,抽滤,旋干滤液,丙酮洗,抽滤,混合滤饼干燥得12. 4 g,收率81.0%.熔点102 ~103 ℃.1H NMR(CDCl3,300 MHz)δ(ppm):1.85 ~2.01(m,2H,CH2);3.67(t,J =4.0 Hz,2H,CH2Cl);3.83(s,3H,CH3);4. 05(t,J =6.0 Hz,2H,CH2O);7.46(s,1H,ArH);7.66(s,1H,ArH);7.53 ~7.77(m,4H,ArH);9.18(s,1H,ArH).
4)4 -(间氯苯胺基)-7 -甲氧基-6 -(3 -氯丙氧基)喹唑啉(3b)的合成.
向100 mL 茄形瓶中,加入50 mL 异丙醇、4 -氯-7 -甲氧基-6 -(3 -氯丙氧基)喹唑啉1(10.0 g,35. 0 mmol)和 间 氯 苯 胺(5. 1 g,40. 0 mmol),加完后回流反应30 min,TLC 监测反应完毕,抽滤,滤饼用叔丁醇洗,干燥得类白色固体11.242g,收率85. 3%.熔点110 ~113 ℃.熔点108 ~109 ℃.1H NMR(CDCl3,300 MHz)δ(ppm):1.85 ~2.01(m,2H,CH2);3.67(t,J =4.0 Hz,2H,CH2Cl);3. 83(s,3H,CH3);4. 05(t,J =6. 0 Hz,2H,CH2O);7.46(s,1H,ArH);7.51(s,1H,NH);7.66(s,1H,ArH);6.99 ~7.53(m,4H,ArH);8.51(s,1H,ArH).
3.2.2 新化合物的合成
1)4 -(对溴苯硫醚基)-7 -甲氧基-6 -(3 -(4 -四氢吡咯基)丙氧基)喹唑啉(4a)的制备.
向50 mL茄形瓶中,加入20 mL DMF、4 -(对溴苯硫醚基)-7 -甲氧基-6 -(3 -氯丙氧基)喹唑啉3a(1.0 g,23 mmol)、无水碳酸钾(0.192 g,28 mol)和碘化钾(0.382 4 g,23 mmol),室温下搅拌10 min,再缓慢滴加四氢吡咯(0.178 2 g,0.002 5 mmol),加毕,75 ℃反应2.5 h,TLC监测反应完毕.反应液冷却后加入20 mL 冰水中,二氯甲烷20 mL萃取3 次,合并有机相,无水硫酸钠干燥,旋干,加入少量乙醚,滴加盐酸-乙醇,慢慢产生类白色固体,抽滤,干燥得产品0. 972 g,收率90.1%.(数据)1H NMR(CDCl3,300 MHz)δ(ppm):1. 69(t,J =4.2 Hz,4H,CH2);1.85(m,2H,CH2);2.46 ~2.51(m,6H,CH2);7.47(s,1H,ArH);7.63(s,1H,ArH);7.53 ~7. 77(m,4H,ArH);9. 18(s,1H,ArH).
2)4 -(对溴苯硫醚基)-7 -甲氧基-6 -(3 -(4 -吗啉基)丙氧基)喹唑啉(4b)的制备.
向50 mL茄形瓶中,加入20 mL DMF、4 -(对溴苯硫醚基)-7 -甲氧基-6 -(3 -氯丙氧基)喹唑啉3a(1.0 g,2.3 mmol)、无水碳酸钾(0.392 4 g,2.8 mmol)和碘化钾(0.382 4 g,2.3 mmol),室温搅拌10 min,再缓慢滴加吗啉0.218 g(2.5 mmol),加毕,75 ℃反应2.5 h,TLC 监测反应完毕.反应液冷却后加入20 mL 冰水中,二氯甲烷(3 ×20 mL)萃取,合并有机相,无水硫酸钠干燥,旋干,加入少量乙醚,滴加盐酸-乙醇,慢慢产生类白色固体,抽滤,干燥得产品1.051 g.收率94.1%.熔点123 ~124 ℃.熔点527. 1 ~529. 5.1HNMR(CDCl3,300 MHz)δ(ppm):1.82(m,2H,CH2);2.32 ~2.49(t,J=6. 5 Hz,6H,CH2);3. 56(t,J =6. 0 Hz,4H,CH2);3.83(s,1H,CH3);4.06(t,J =6.5 Hz,2H,CH2);7.46(s,1H,ArH);7.53(d,J =8.5 Hz,2H,ArH,ArH);7.64(s,1H,ArH);7.77(dm,2H,ArH,ArH);9.16(s,1H,ArH).
3)4 -(3 -氯苯胺基)-7 -甲氧基-6 -(3 -(N-甲基-5 -甲基苯并咪唑基巯基)丙氧基)喹唑啉(4c)的制备.
向50 mL茄形瓶中,加入20 mL DMF、4 -(3 -氯苯胺基)-7 -甲氧基-6 -(3 -氯丙氧基)喹唑啉3b(1.0 g,2.6 mmol)、无水碳酸钾(0.4 g,3.2 mmol)和碘化钾(0.4 g,2.6 mmol),室温搅拌10 min,再缓慢加入N -甲基-2 -巯基-5 -甲基苯并咪唑(0.5 g,2.7 mmol),75 ℃反应4 h,TLC监测反应完毕.反应液冷却后加入30 mL 冰水中,有大量白色固体产生,抽滤,干燥得产品1. 3 g.收率98.4%.熔点150 ~152 ℃.1H NMR(CDCl3,300 MHz)δ(ppm):2.11(m,2H);3.10(t,J =6.5 Hz,2H);3. 72(s,1H);3. 83(s,1H);4. 0(brs,1H,NH);4. 06(t,J =6. 0 Hz,2H);6. 81 ~6. 85(m,2H);7.14(t,1H);7.24(s,1H);7.41(s,1H);7.52(m,1H);7. 82(s,1H);7. 93(m,1H);8. 49(s,1H).
4)4 -(3 -氯苯胺基)-7 -甲氧基-6 -(3 -(N -甲基-5 -三氟甲基苯并咪唑基巯基)丙氧基)喹唑啉(4d)的制备.
向50 mL茄形瓶中,加入20 mL DMF、4 -(3 -氯苯胺基)-7 -甲氧基-6 -(3 -氯丙氧基)喹唑啉3b(1. 0 g,2. 6 mmol)、无水碳酸钾(0. 4 g,3.2 mmol)和碘化钾(0.4 g,2.6 mmol),室温搅拌10 min,再缓慢加入N-甲基-2 -巯基-5 -三氟甲基苯并咪唑(0.6 g,2.7 mmol),75 ℃反应6 h,TLC监测反应完毕.反应液冷却后加入30 mL冰水中,有大量白色固体产生,抽滤,干燥得产品1.5 g.收率98.5%.熔点163 ~164 ℃.1H NMR(CDCl3,300 MHz)δ(ppm):2.11(m,2H);3.10(t,J =6.0 Hz,2H);3.72(s,1H);3.83(s,1H);4.0(brs,1H,N-H);4. 06(m,2H);6. 81 ~6. 85(m,2H);7. 14(m,1H);7. 24(s,1H);7. 41(s,1H);7. 52(m,1H);7.82(s,1H);7.93(m,1H);8.49(s,1H).
5)4 -(对溴苯亚砜基)-7 -甲氧基-6 -(3 -(4 -四氢吡咯基)丙氧基)喹唑啉(4e)的制备.
向50 mL 茄形瓶中,加入20 mL 冰乙酸、4 -(对溴苯硫醚基)-7 -甲氧基-6 -(3 -(4 -四氢吡咯基)丙氧基)喹唑啉4a(0.5 g,1.0 mmol)和过硼酸钠(0.179 2 g,12 mmol),加毕,室温反应45 min,TLC 监测反应完毕.反应液用氢氧化钠调将pH值调至10,静置,有固体析出,抽滤,干燥得类白色固体0.481 5 g,收率98.1%,熔点135 ~137 ℃.1H NMR(CDCl3,300 MHz)δ(ppm):1.68(m,4H,CH2);1. 85(m,2H,CH2);2.46 ~2. 51(m,6H,CH2);3.83(s,1H,CH3);4.06(t,J =4.0 Hz,2H,CH2);7.38(d,2H,ArH);7.47(s,1H,ArH);7.64(s,1H,ArH);7.78(d,J =16.5 Hz,2H,ArH);9.50(s,1H,ArH).
6)4 -(对溴苯亚砜基)-7 -甲氧基-6 -(3 -(4 -吗啉基)丙氧基)喹唑啉(4f)的制备.
向50 mL 茄形瓶中,加入20 mL 冰乙酸、4 -(对溴苯硫醚基)-7 -甲氧基-6 -(3 -(4 -吗啉基)丙氧基)喹唑啉4b(0.5 g,1.0 mmol)和过硼酸钠(0.173 2 g,1.1 mmol),加毕,室温反应45 min,TLC监测反应完毕.反应液用氢氧化钠调将pH 值调至10,静置,有固体析出,抽滤,干燥得类白色固体0. 486 5 g.收率96. 8%.熔点129 ~130 ℃.1H NMR(CDCl3,300 MHz)δ(ppm):1.68(m,4H);1.82(m,2H);2.46 ~2.51(m,6H);3.83(s,1H);4.06(t,J =6.0 Hz,2H);7.24(s,1H);7.41 ~7.47(m,3H);7.82(dm,2H);9.58(s,1H).
