加速溶剂萃取-QuEChERS-超高效液相色谱-串联质谱法测定药食同源性食品中16种真菌毒素
2020-05-22古淑青陈柔含邓晓军郭德华
方 真, 曲 栗, 古淑青*, 陈柔含, 李 优, 邓晓军, 郭德华, 冯 峰
(1. 上海海关动植物与食品检验检疫技术中心, 上海 200135; 2. 上海大学生命科学学院食品工程系, 上海 200436; 3. 中国检验检疫科学研究院, 北京 100176)
真菌毒素是一种由产毒丝状真菌在一定环境条件下产生的具有毒性作用的次级代谢产物[1],易污染农产品、食品和中药材等植物源性产品。目前已知的真菌毒素超过300多种,其中大约有30种易对人体健康产生严重威胁,包括黄曲霉毒素(AFT)、赭曲霉毒素A(OTA)和脱氧雪腐镰刀菌烯醇(DON)等,过量摄入受污染的食品会导致肝肾功能损坏、致癌、致畸,或诱发免疫抑制性疾病[2,3]。随着真菌毒素风险评估工作的不断深入,许多地区和国家都对真菌毒素的含量进行了强制性规定。我国GB 2761-2017《食品安全国家标准食品中真菌毒素限量》中对上述多种真菌毒素最为严格的限量为5 μg/kg,而欧盟等国家则为2 μg/kg。
目前食品中真菌毒素的检测方法主要有以酶联免疫吸附测定法[4]和薄层色谱法[5]为主的定性和半定量检测方法,以及以毛细管电泳法[6]、液相色谱法[7]为主的定量分析检测方法。这些传统检测方法操作简单,成本低廉,但定性干扰大,定量灵敏度不足,且只能一次实现一种或一类真菌毒素的定性和定量分析,无法满足实际样品中多种痕量真菌毒素污染物同时检测的需求。近年来,随着质谱技术的快速发展,高效液相色谱-串联质谱法(HPLC-MS/MS)在检测毒素方面的优势逐渐显现[8]。HPLC-MS/MS结合了色谱的分离能力与质谱的分子确证功能,兼具高灵敏与高特异性的优点,是同时检测多种毒素的理想方法,近年来被广泛应用于不同样品中真菌毒素的定性、定量分析[8,9]。
在基于HPLC-MS/MS的检测工作中,样品前处理是关键环节。真菌毒素检测的前处理过程通常包括萃取和净化两个步骤。加速溶剂萃取(accelerated solvent extraction, ASE)是近年来新兴的一种萃取技术,在较高的温度和压力下,利用一定配比的有机溶剂对固体样品进行萃取[10]。ASE不仅能够提高目标化合物的萃取效率,而且能够减少萃取溶剂的使用量,缩短萃取时间。QuEChERS净化方法是近年来最新发展起来的一种主要用于农残检测的前处理方法[11],选择性使用多种混合净化填料组合可以高效地去除多种杂质。QuEChERS具备高效、简便、绿色等优点,近年来广泛应用于各检测领域,也包括真菌毒素的分析[12-14]。将ASE和QuEChERS联合使用,既能够提高目标化合物的萃取效率,又满足了前处理操作简便、快速的要求[15]。但是,目前还未见结合两种技术应用于药食同源产品中多种真菌毒素检测的报道。
本实验采用加速溶剂萃取结合QuEChERS方法对药食同源样品中16种真菌毒素进行萃取和净化,采用超高效液相色谱-串联质谱技术进行测定。该方法操作简单,重复性好,灵敏度高,可用于药食同源产品中多种真菌毒素的快速筛查和定量检测。
1 实验部分
1.1 仪器、试剂与材料
Nexera X2液相色谱仪(日本Shimadzu公司); QTrap 6500三重四极杆-线性离子阱复合质谱系统(美国AB Sciex公司); Allegra X-22R离心机(美国Beckman Coulter公司); ASE 350加速溶剂萃取仪(美国Thermo Fisher公司); R-215旋转蒸发仪(瑞士Buchi公司); N-EVAPTM112氮吹仪(美国Organomation Associates公司)。所有实验室用水均由Milli-Q超纯水系统(美国Millipore公司)制得。
16种真菌毒素标准品:DON(CAS号:51481-10-8,纯度≥99.0% )、玉米赤霉烯酮(ZEN, CAS号:17924-92-4,纯度≥99.9% )、3-乙酰基脱氧雪腐镰刀菌烯醇(3-AcDON, CAS号:50722-38-8,纯度≥99.0% )、15-乙酰基脱氧雪腐镰刀菌烯醇(15-AcDON, CAS号:88337-96-6,纯度≥99.0% )、黄曲霉毒素B1(AFB1, CAS号:1162-65-8,纯度≥99.0% )、黄曲霉毒素B2(AFB2, CAS号:7220-81-7,纯度≥99.9% )、黄曲霉毒素G1(AFG1, CAS号:1165-65-8,纯度≥99.0% )、黄曲霉毒素G2(AFG2, CAS号:7241-98-7,纯度≥98.3% )、伏马毒素B1(FB1, CAS号:116355-83-0,纯度≥99.7% )、伏马毒素B2(FB2, CAS号:116355-84-1,纯度≥99.9% )、OTA(CAS号:303-47-9,纯度≥99.9% )、赭曲霉毒素B(OTB, CAS号:4825-86-9,纯度≥99.0% )、疣孢青霉原(VER, CAS号:12771-72-1,纯度≥98.0% )、杂色曲霉毒素(SMC, CAS号:10048-13-2,纯度≥99.9% )、T-2毒素(T-2, CAS号:21259-20-1,纯度≥98.2% )、HT-2毒素(HT-2, CAS号:26934-87-2,纯度≥99.0% )、黄曲霉毒素B1内标(U-[13C17]-AFB1, CAS号:1217449-45-0, 0.5 mg/L)、伏马毒素B1内标(U-[13C34]-FB1, CAS号:1217458-62-2, 25 mg/L)均购自美国Sigma公司;胺丙基键合硅胶(-NH2)、乙二胺-N-丙基硅烷(PSA)、C18均购自上海安谱实验科技股份有限公司;甲酸(色谱纯,美国Fluka公司);乙酸(分析纯,国药集团化学试剂有限公司);乙腈、甲醇(色谱纯,美国Thermo Fisher公司)。山银花、葛根、沙棘样品均为市售。
1.2 混合标准溶液配制
混合标准储备液:分别准确称取适量各标准品,用乙腈-甲醇(3∶2, v/v)配制成含各真菌毒素100 mg/L 的混合标准储备液,于-20 ℃下避光保存。
内标工作液:用乙腈将U-[13C17]-AFB1配制成质量浓度为100 μg/L 的同位素内标工作液,U-[13C34]-FB1配制成质量浓度为250 μg/L 的同位素内标工作液,于-20 ℃下避光保存。
基质混合标准工作溶液:用空白样品萃取液将混合标准储备液配制成适当浓度的基质混合标准工作溶液,并加入同位素内标溶液,现用现配。
1.3 样品前处理
1.3.1 萃取
准确称取粉碎过筛后的样品5.00 g,加入黄曲霉素B1同位素内标工作液和伏马毒素B1同位素内标工作液各400 μL,然后加入1 g氯化钠,并与适量硅藻土混匀后,移入34 mL不锈钢样品萃取池中进行加速溶剂萃取。萃取试剂为乙腈-水-乙酸(85∶12∶3, v/v/v),萃取温度75 ℃,加热时间5 min,静态萃取时间5 min,循环2次,冲洗体积为60%萃取池体积,萃取后氮气吹扫100 s,收集萃取液于60 mL收集瓶中。最后将萃取液转移至鸡心瓶中,于45 ℃水浴中减压浓缩至近干,加入20 mL乙腈-水(85∶15, v/v)充分溶解残渣,待净化。
1.3.2 净化
取上述待溶液5 mL于离心管中,加入0.6 g -NH2、0.4 g PSA、0.25 g C18,涡旋振荡5 min。以 10 000 r/min 离心5 min,吸取1 mL上清液于室温氮吹至近干,加入1 mL含0.1%(v/v)甲酸的乙腈-水(3∶97, v/v)复溶,过0.22 μm微孔滤膜,待上机。
1.4 分析条件
反相色谱柱:Kinetex XB-C18柱 (100 mm×3 mm, 2.6 μm,美国Phenomenex公司);柱温:40 ℃;流动相A为0.1%(v/v)甲酸水溶液(含5 mmol/L 乙酸铵), B为0.1%(v/v)甲酸乙腈溶液;流速:0.4 mL/min。梯度洗脱条件:0~0.5 min, 3%B; 0.5~1.0 min, 3%B~10%B; 1.0~6.0 min, 10%B~90%B; 6.0~7.5 min, 90%B; 7.5~7.6 min, 90%B~3%B; 7.6~9.0 min, 3%B。进样体积:10 μL。
离子源:电喷雾电离(ESI)源,采用正、负离子同时扫描模式;喷嘴电压:5 500 V(+)/-4 500 V(-);毛细管温度:600 ℃;气帘气压力0.207 MPa;碰撞气电压强度:medium。16种真菌毒素的质谱参数见表1。
2 结果与讨论
2.1 前处理条件优化
2.1.1 ASE条件优化
16种真菌毒素的理化性质差异较大,因此选择一种兼容性好、萃取效率高的萃取溶剂尤为重要。本实验在现有文献[16-18]报道的基础上,分别用乙腈、乙腈-乙酸体系和乙腈-水-乙酸体系作为萃取溶剂对空白山银花样品进行加标试验(见图1)。在pH值相同的情况下,当萃取试剂中含有适量水时,大部分真菌毒素的萃取效率相比使用纯乙腈时均有提高,这是因为水能够增强乙腈的渗透能力,以便于萃取液渗透到基质较为复杂的药食同源性食品中,从而提高目标物的萃取效率。此外,pH值对于部分酸敏感真菌毒素的萃取效率有较大影响,如DON、ZEN、OTA、FB1、FB2具有羧基基团,水溶性强,对有机萃取溶剂的酸度有一定要求,因此降低萃取液的pH值能提高其稳定性,增加萃取效率。实验进一步对比了当萃取液中含0% 、1% 、3% 和5%体积分数的乙酸时的萃取效率。从图1可以看出,随着乙酸体积分数的增加,部分真菌毒素的萃取效率有所增加,而当萃取液中含有3%的乙酸时各目标化合物的萃取效率相对较好。因此,实验选择乙腈-水-乙酸(85∶12∶3, v/v/v)作为萃取试剂。

表 1 16种真菌毒素及两种同位素内标的质谱参数Table 1 MS parameters of the 16 mycotoxins and two isotopic internal standards
CE: collision energy; DP: declustering potential; * quantitative ion.
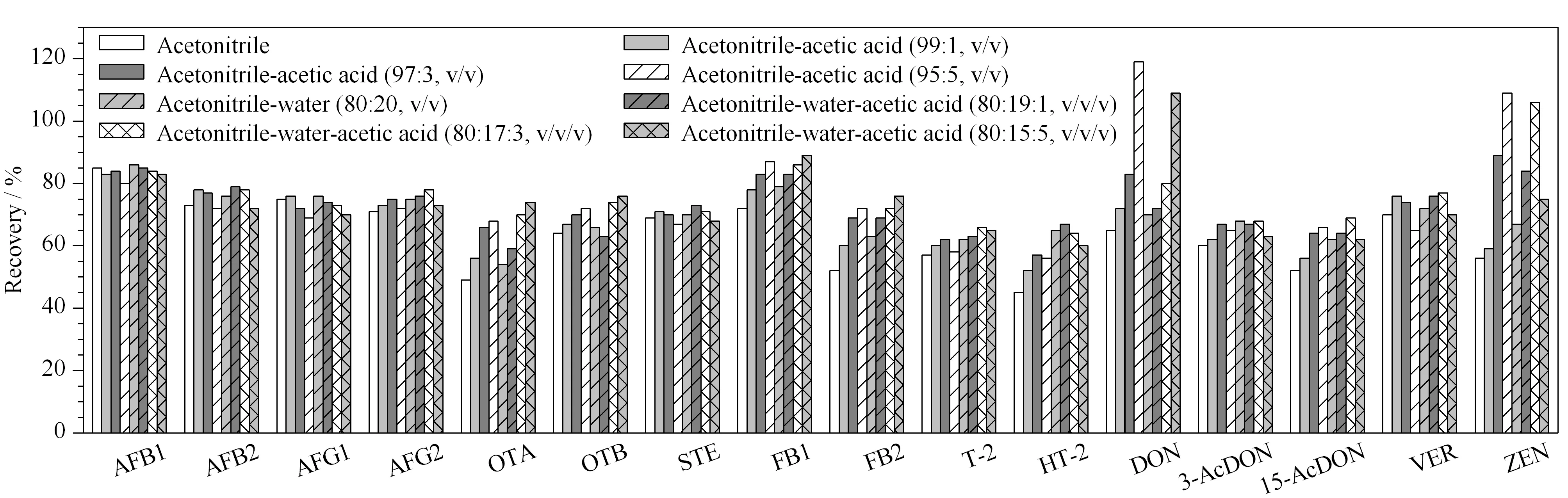
图 1 萃取液对加标山银花样品中16种真菌毒素萃取效率的影响Fig. 1 Effect of extraction solvents on the extraction efficiencies of the 16 mycotoxins spiked in the flos lonicerae samples
萃取温度考察了55、75、85和95 ℃ 4个温度点。温度增加可以降低萃取溶剂的黏度,从而提高溶剂对基质的浸润能力和对目标物的溶解能力。但是温度过高也可能导致目标物产生不可预知的变化,从而影响最终的萃取效果。结果表明,当萃取温度为85 ℃和95 ℃时,各类毒素的回收率都有所减少。综合考率,本实验选择75 ℃作为萃取温度。
在循环次数上,循环2次与循环3次,结果没有明显的差异,为了减少样品前处理的时间,选择循环2次。
整个萃取过程大约用时20 min,得到的萃取液较为清澈,无需过滤直接进行蒸发浓缩。
2.1.2 净化填料的优化
药食同源性食品基质较为复杂,且实验采用高温高压萃取方式,在萃取目标物化合物的同时也会萃取样品中其他干扰物,如蛋白质、色素、脂质和糖类等,因此净化步骤对于结果的准确性至关重要。净化剂可以通过极性相互作用或非极性相互作用使目标化合物和杂质相互分离。本研究考察的净化剂主要有C18、PSA、石墨化炭黑(GCB)、-NH2、氰丙基键合硅胶(-CN)、弗洛里硅土(Florisil)。在空白山银花基质中进行添加回收试验,各分析物的添加水平为10 μg/kg。分别考察了采用6种净化剂单独处理以及不同配比的净化剂组合处理的净化效果,比较各真菌毒素的回收率。结果表明,使用GCB时,黄曲霉毒素、玉米赤霉烯酮回收率较低。因为GCB对具有平面分子结构的杂质具有显著的吸附作用[19],但目标化合物中黄曲霉毒素等毒素具有平面结构或不完全平面分子结构,因此GCB不适于本研究。含有氰丙基键合硅胶和胺丙基键合硅胶等活性基团的净化剂可通过氢键作用吸附极性物质(脂肪酸和有机酸等),实验中发现-NH2能强烈吸附基质中的极性色素,净化非极性毒素。C18能够吸附基质中非极性物质,有效去除糖类,但净化能力有限,加入PSA和-NH2后,各类真菌毒素的回收率都得到了提高,这与糖类、有机酸、脂肪酸等主要干扰物质被去除后降低了基质效应的影响有关[20]。因此,本实验最终采用C18、PSA和-NH2协同净化的方式,增强杂质的净化能力,降低离子抑制作用,提高方法的准确度。
2.2 超高效液相色谱条件优化
实验考察了2种色谱柱(Waters, ACQUITY UPLC HSS T3 (100 mm×2.1 mm, 1.8 μm)和Phenomenex, Kinetex XB-C18 (100 mm×3 mm, 2.6 μm))对16种真菌毒素分离效果的影响。结果表明:使用T3柱时,3-AcDON和15-AcDON这对同分异构体无法达到基线分离,且峰形较差,响应较低。而使用Phenomenex Kinetex XB-C18柱则能达到较好的分离效果。该柱内径和填料粒径均较小,能够获得良好的分离度。
由于真菌毒素易溶于甲醇或乙腈,实验还分别考察了甲醇和乙腈作为强洗脱流动相的洗脱效果。当乙腈-水体系作为流动相时,在正、负两种模式下的洗脱效果和响应强度均好于甲醇-水体系,这有可能是因为少数待测物中含有乙酰氧基团,而此类化合物在甲醇中不稳定。因此,实验选择乙腈作为强洗脱流动相。在流动相中加入0.1%(v/v)的甲酸可以提高待测物在电喷雾电离、正离子模式中的离子化效率,提高灵敏度。当使用0.1%(v/v)甲酸水和0.1%(v/v)甲酸乙腈进行梯度洗脱时,AFG2存在严重的拖尾现象,而在水相中加入少量的乙酸铵能消除这种现象。为了增大目标化合物的响应值并且改善峰形,实验进一步考察了不同浓度的乙酸铵对目标化合物离子化效率的影响。结果表明,在水相中加入5 mmol/L 乙酸铵能够最大限度增加目标化合物的响应值并改善峰形,而过高浓度的乙酸铵反而会降低目标化合物的离子化效率。所以实验最终选择含5 mmol/L 乙酸铵的0.1%(v/v)甲酸水溶液和0.1%(v/v)甲酸乙腈溶液作为流动相。由于药食同源性食品基质较为复杂且分析物较多,实验选择梯度洗脱进行分析以达到缩短出峰时间和高效除去色谱柱中残留杂质的目的。
2.3 质谱条件优化
在优化质谱采集参数时,同时考察了16种真菌毒素在正、负离子模式下的响应值,发现除了ZEN在负模式下响应较好,其余的15种目标物均在正离子模式下响应较好。除了T-2的母离子为[M+NH4]+峰,其余15种化合物在选定的分析条件下都产生[M+H]+峰。在MRM分析中选择了离子丰度最高的子离子作为定量离子,离子丰度次高的作为定性离子,并且优化了CE值和DP值,以获得较高的响应值。各化合物的提取离子流色谱图见图2。

图 2 16种真菌毒素标准品及同位素内标的提取离子流色谱图Fig. 2 Extracted ion current chromatograms of the 16 mycotoxins and the isotope internal standards
2.4 方法学验证
2.4.1 标准曲线、线性范围及检出限
由于AFB1和FB1的稳定同位素内标试剂易获得,实验中AFB1和FB1用内标法定量,其余毒素用外标法定量。
在空白山银花样品基质中添加目标化合物,分别加入100 μL AFB1和FB1的同位素内标工作液,配制成系列浓度的标准工作溶液。AFB1和FB1以其与同位素内标响应峰面积之比为纵坐标(Y′),其余14种化合物以峰面积(Y)为纵坐标,分别以对应的质量浓度(X, μg/L)为横坐标,绘制标准曲线。以3倍和10倍信噪比(S/N)确定方法的检出限(LOD)和定量限(LOQ)。
如表2所示,16种真菌毒素在各自的线性范围内线性良好,线性相关系数(r2)均大于0.99。16种真菌毒素的检出限为0.008~0.3 μg/kg,定量限为0.03~1.0 μg/kg,远低于国家标准对真菌毒素限量检测要求和欧盟、日本等国家规定的最大残留量。
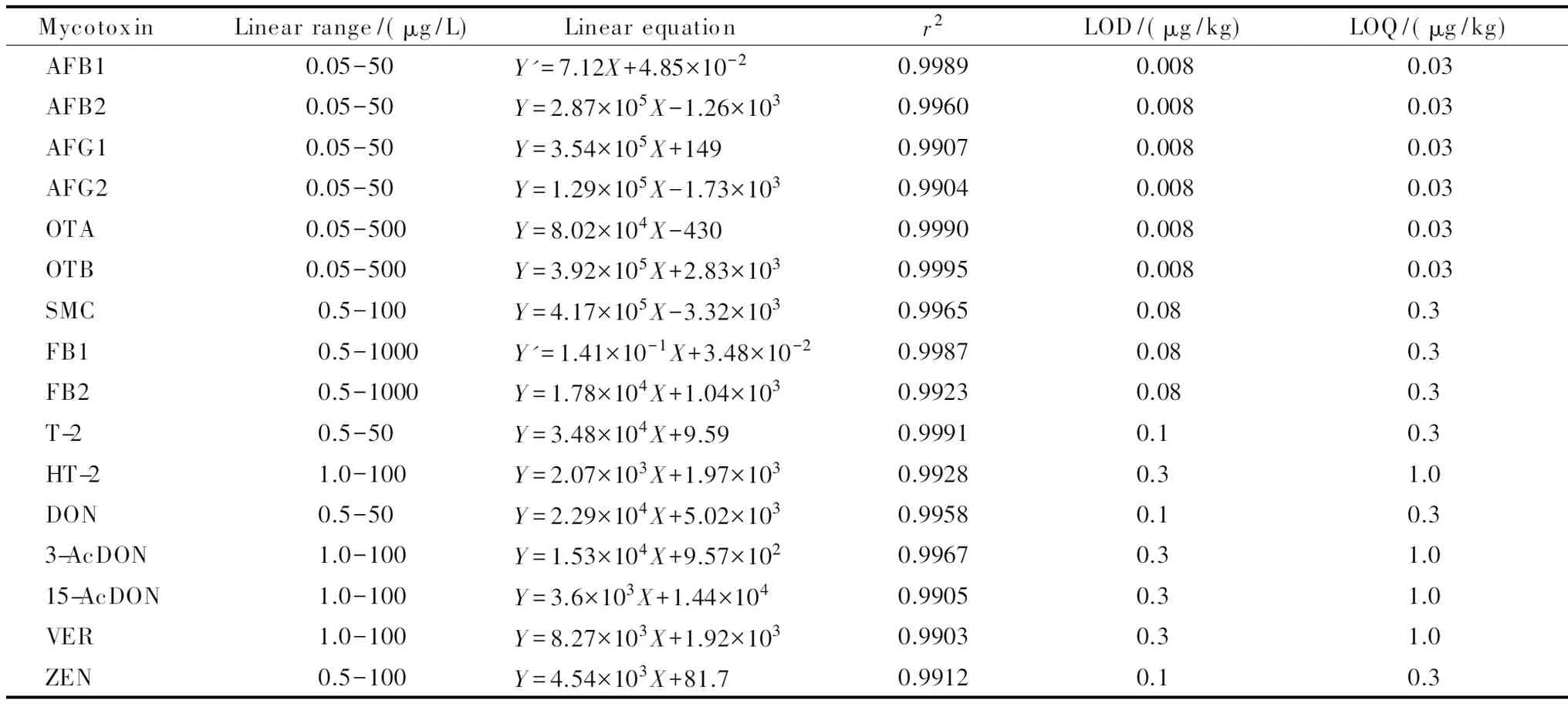
表 2 16种真菌毒素的线性范围、线性方程、相关系数、检出限和定量限Table 2 Linear ranges, linear equations, correlation coefficients (r2), LODs and LOQs of the 16 mycotoxins
Y: peak area;Y′: peak area ratio of the analyte to isotope internal standard;X: mass concentration, μg/L.
2.4.2 加标回收率和精密度
分别用山银花、葛根、沙棘空白样品作为基质,添加3个不同水平的待测物,平行测定6次,计算回收率以及相对标准偏差(RSD),结果见表3。可以看出,16种真菌毒素的平均回收率为70.8% ~118% ,使用内标校准后,AFB1和FB1的回收率更高,可达90%以上;RSD为2.5% ~10.2% ,满足检测要求。
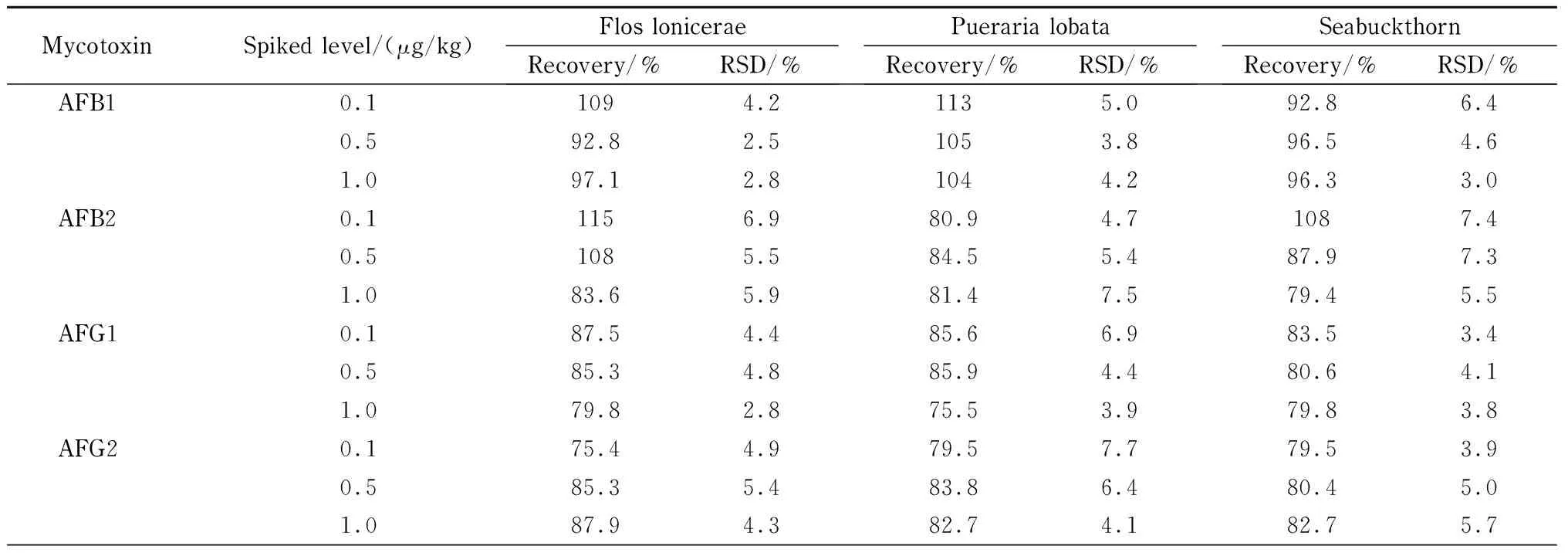
表 3 16种真菌毒素在3种基质中的回收率及相对标准偏差(n=6)Table 3 Recoveries and relative standard deviation (RSDs) of the 16 mycotoxins spiked in three matrices (n=6)

表 3 (续)Table 3 (Continued)
2.5 实际样品检测
采用建立的方法分别对电商销售的30个批次的山银花、葛根和沙棘进行16种真菌毒素的残留量测定。
结果表明,1个批次的沙棘样品和1个批次的葛根样品被检出含有AFB1,污染水平分别为3.59 μg/kg 和6.17 μg/kg。1个批次的沙棘样品被检出含有AFG2,污染水平高达11.7 μg/kg。1个批次的山银花样品被检出含有OTA,污染水平为7.45 μg/kg。2个批次的沙棘样品和1个批次的葛根样品被检出含有FB1,污染水平为52.6~96.9 μg/kg。其余真菌毒素均未检出。
3 结论
本文建立了一种基于ASE和QuEChERS联用的前处理技术,结合超高效液相色谱-串联质谱同时检测药食同源性食品中16种真菌毒素的检测方法。与传统方法相比,本方法减少了有机溶剂用量,缩短了检测周期,提高了检测灵敏度,检出限、定量限远低于国家标准对真菌毒素限量检测要求和欧盟、日本等国家规定的最大残留量。方法学考察及实际样品检测证明,该方法具有实用性强、灵敏度高、操作简便快速等优点,能够满足国内外对植物源性药食同源产品中多种真菌毒素的检测要求。
