高效液相色谱法测定饮用水中草甘膦的不确定度评定
2020-05-21余秀娟樊继鹏王伟伟魏晓培顾娟红
余秀娟 樊继鹏 王伟伟 魏晓培 顾娟红
(1.苏州海关综合技术中心,江苏 苏州 215104;2. 苏州华博日化品检测服务有限公司,江苏 苏州 215104)
草甘膦,化学名为N-(膦酰基甲基)甘氨酸,化学分子式为C3H8NO5P,又称为镇草宁、农达、草干膦、膦甘酸,属于弱有机酸,是一种常用的水溶性除草剂,纯品为非挥发性白色固体,不溶于一般有机溶剂,不可燃,不爆炸,常温下可以稳定贮存。草甘膦为内吸传导性强的广谱灭生性除草剂,在农业上大量使用的草甘膦除草剂,只有极少部分发挥了作用,多数残留在土壤或漂浮在大气中,通过降雨和径流的冲刷进入地表水或地下水,造成水体污染。随着草甘膦使用频率不断上升,其残留问题日益受到关注。长期饮用含有草甘膦农药的水,会危及人体健康,并会造成肝肾、消化道黏膜、皮肤黏膜、神经系统、呼吸系统和心血管系统的损伤,严重者可危及生命。因此,草甘膦的检测技术一直是草甘膦污染防治关注的焦点。目前草甘膦的测定方法主要有气相色谱法、气相色谱质谱法、高效液相色谱法、液相色谱-质谱联用法、分光光度法、酶联免疫法、毛细管电泳法、离子色谱法等,其中最常用的方法是高效液相色谱法[1-3]。为了快速、准确测定生活饮用水中痕量草甘膦,保证生活饮用水卫生安全、保护人体健康,本文建立超高效液相色谱-柱后衍生法测定饮用水中草甘膦的检测方法,对所建立方法测定草甘膦的不确定度进行了评定。
1 材料和方法
1.1 主要仪器和设备
LC 1260 液相色谱仪,配有自动进样器(美国 Agilent 公司);色谱柱:Potassium Cation Exchange柱(150 mm×4.0 mm,8 μm,PICKING公司) ;Milli-型超纯水机(Merck Millipore中国有限公司);PVDF 针式滤器:25 mm×0.22 μm(上海安谱实验科技股份有限公司)。
1.2 试剂
草甘膦标准溶液:100 μg/mL(北京坛墨质检科技有限公司);甲醇:HPLC级,纯度不低于99.9(德国Merck公司);K200(0.4%H3PO4、0.1%KH2PO4、99.5%H2O)、RG019(0.3%KOH、99.7%H2O)、GA116(0.1%NaOH、0.1% KH2PO4、1% NaCl、98.8%H2O)、GA104(5.4%KBO3、94.6%H2O)、邻苯二醛、2-巯基乙醇、草甘膦恢复液(4.1% H3PO4、0.3% KH2PO4、95.6%H2O);10.0%次氯酸钠(PICKING公司);5%次氯酸钠(国药集团);实验用水为纯水。
1.3 分析方法
1.3.1 柱后衍生仪器条件
柱后衍生反应器温度:36℃;泵流速:0.30 mL/min;衍生剂1:吸取100 μL溶于1000 mL稀释剂GA116中。衍生剂2:称取100 mg 邻苯二醛溶于10 mL甲醇中,称取2.0g 2-巯基乙醇溶于5 mL稀释剂GA104中,然后将二者全部溶于1000 mL稀释剂GA104中,通氮气脱气10 min。
1.3.2 液相色谱仪器条件
柱温:55℃;流量:0.40 mL/min;进样体积:20 μL;流动相:通道A=K200,100.0%,通道B=RG019,0.00%;梯度洗脱程序:初始 A:B(体积比)=100:0;15.1 min 时,A:B(体积比)=0:100;17.1 min 时,A:B(体积比)=100:0;检测器:FLD;检测波长:激发波长Ex=330 nm,发射波长Em=465 nm。
1.4 不确定度来源分析及数学模型的建立
依据JJF 1059.1-2012[4],本方法的不确定度来源主要有:B类不确定度:(1)标准溶液引入的不确定度urel(1);(2)标准曲线回归引入的不确定度urel(2)。A类不确定度:(3)重复性测定产生的不确定度urel(3)。对这些来源产生的不确定度进行量化,本方法的不确定度数学模型为:
2 结果与讨论
2.1 标准曲线的绘制
准确吸取1.0 mL 草甘膦标准溶液于10.0 mL容量瓶中,用水稀释至刻度,配制成浓度为10.0 μg/mL的标准使用液。稀释标准使用液配制成浓度为 0.025、0.05、0.10、0.50、1.00、2.00 μg/mL的标准曲线,按照1.3中的仪器条件进行测定,外标法定量。线性范围为0~2.0 μg/mL,线性回归方程为Y=27.10205*X-0.150174,相关系数0.9999,保留时间8.602 min(图1)。
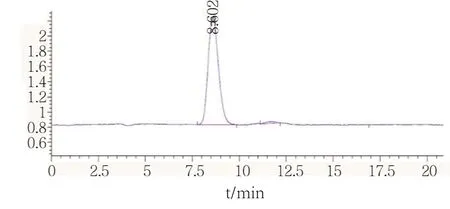
图1 标准样品谱图
2.2 准确度和精密度试验
用阴性自来水样品分别添加标准溶液配制成0.10、0.40、2.00 μg/mL三个浓度水平的样品,每个水平进行6次重复试验,结果见表1。平均回收率在98.0%~105.0%之间,相对标准偏差在0.7%~5.5%,以3倍信噪比(S/N=3)确定方法的检出限,本方法检出限为0.025 μg/mL。该方法检出限满足生活饮用水中草甘膦的日常检测要求。
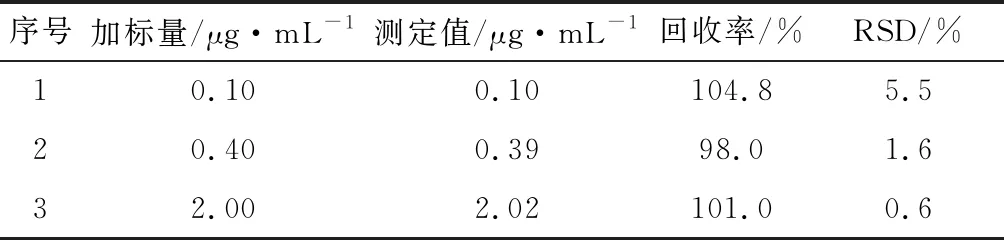
表1 回收率和精密度试验结果
2.3 样品分析
用棕色磨口玻璃瓶采集水样,采样时水样充满玻璃瓶。当余氯存在时,加入硫代硫酸钠,使其在水中的浓度达到100 μg/mL。水样采集后,将样品溶液于4℃避光保存,并在两周内测定。分析时,水源水需30000转/min的转速下离心5min, 取上清液用PVDF针式滤器将样品溶液过滤,同标准曲线一起上机测试;出厂水、管网水等清洁水直接用PVDF针式滤器将样品溶液过滤,同标准曲线一起上机测试。笔者用所建立的方法分别测定了自来水和水源水样品各10个,均未检测到草甘膦。
2.4 不确定度评定
2.4.1 标准溶液及配制引入的不确定度urel(1)
草甘膦标准溶液浓度为100 μg/mL,测定的标准不确定度为3.0 μg/mL,则相对标准不确定度分量为:u(1)=3.0/100=0.03。配制标准曲线时,标准使用液为10.0 μg/mL,稀释10倍使用,则urel(1)=0.03/10=0.003。
2.4.2 标准曲线回归引入的不确定度urel(2)
草甘膦的标准曲线测定结果见表2。实测值与理论值一一对应关系见表3。

表2 草甘膦的标准曲线测定结果
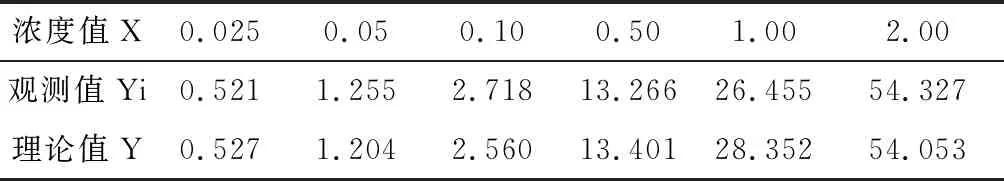
表3 实测值与理论值对应关系
配制浓度为0.1 μg/mL的标准样品平行测定6次,响应值分别为2.4024、2.7624、2.8335、2.7177、2.6327、2.7919,代入标准曲线得到的浓度分别0.094 μg /mL、0.107 μg /mL、0.110 μg /mL、0.105 μg /mL、0.103 μg/mL、0.108 μg/mL,其标准不确定度为:
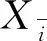
草甘膦标准曲线拟合产生的相对标准不确定度为:urel(2)= u(2)/0.105=0.3905。
2.4.3 重复性测定产生的不确定度urel(3)
实验室人员操作的重复性、仪器进样的重复性、数据软件处理的重复性等因素组成了重复实验不确定度的来源[5-8]。本实验室重复测定6次,加标样品测定结果分别为0.094 μg/mL、0.107 μg/mL、0.110 μg/mL、0.105 μg/mL、0.103 μg/mL、0.108 μg/mL,其平均值为0.105 μg/mL,其标准偏差为0.006 μg/mL。
2.4.4 合成标准不确定度
将所有不确定度分量进行合成,则相对合成标准不确定度
合成不确定度u=0.105×0.3912=0.0412 μg/L
取扩展因子k=2,扩展不确定为:U=0.0412×2=0.0824 μg/L,则草甘膦的测定结果应表示为:(0.105±0.0824)μg/L,k=2。
3 结论
本文采用高效液相色谱法测定饮用水中草甘膦,方法操作简便,选择性好,灵敏度高,定量准确,分析时间短。对该方法进行不确定度评定,结果表明B类不确定度中标准曲线回归引入的不确定度影响最大,A类不确定度重复性测定产生的不确定度影响次之,因此应充分重视标准工作溶液的配制,规范各项操作。分析前,将仪器各项参数调试至最佳状态,减小由仪器问题带来的结果偏离。
