迭代饱和突变提高Bacillus cereus胺脱氢酶对苯乙酮还原的催化效率
2020-05-19穆晓清
吴 涛,穆晓清,聂 尧,徐 岩
(1.江南大学工业生物技术教育部重点实验室,无锡 214122;2.宿迁市江南大学产业技术研究院,宿迁 223800)
光学纯(R)-α-苯乙胺是合成治疗阿尔茨海默病药物利凡斯的明(Rivastigmine)的手性砌块[1,2],也是一种有机酸类手性拆分试剂和性能优良的手性助剂[3],在工业上具有广阔的应用前景.(R)-α-苯乙胺的工业化生产大多采用金属催化苯乙酮直接还原胺化[4]、 偶联α-苯乙胺的动力学拆分[5,6]或烯胺的不对称氢化[7]等化学方法.但化学法存在环境污染严重、 副产物多、 产物提纯复杂及催化剂回收困难等缺陷.与化学法合成(R)-α-苯乙胺不同,以脂肪酶[8,9]、 环己胺氧化酶[10]、 转氨酶[11,12]及胺脱氢酶(AmDH)[13,14]为代表的生物合成途径具有反应条件温和、 原料易得及环境友好等优点.其中,脂肪酶和环己胺氧化酶催化的手性拆分需要以外消旋α-苯乙胺为原料,生产成本较高且步骤繁杂; 转氨酶催化的转氨反应需要添加过量的有机胺或氨基酸作为氨基供体,且存在底物抑制和反应平衡的问题,工业应用难度大.采用AmDH催化以潜手性酮和游离氨为底物的不对称还原反应合成手性胺的路线具有绿色高效、 成本低及产物光学纯度高的优点[13~16],是手性胺合成最理想的反应之一.但目前发现的AmDH种类极少且底物谱窄[17,18],严重限制了其工业应用.
2012年,Bommarius等[19]首次以源于Bacillusstearothermophilus的亮氨酸脱氢酶(BsLeuDH)为模板进行活性中心2个高度保守氨基酸的突变(K68S/N261L),将亮氨酸脱氢酶转变成AmDH,开发了手性胺合成的新途径,但制得的BsAmDH对苯乙酮的比活力仅为58.8 U/g(20 mmol/L).Xu等[20]在源于Exiguobacteriumsibiricum的亮氨酸脱氢酶(EsLeuDH)的活性中心引入相同的双突变制得的EsAmDH(K77S/N270L)对苯乙酮的比活力达到98.5 U/g(20 mmol/L,30 ℃).Gröger等[21]以EsAmDH为催化剂合成了(R)-α-苯乙胺,虽然在20,50和100 mmol/L的底物浓度下,产物(R)-α-苯乙胺的对映体过量(e.e.)值可达到99%,但当底物浓度为100 mmol/L时,底物苯乙酮的转化率仅为43%,距离工业应用差距较大.酶对苯乙酮的催化效率低是现阶段限制(R)-α-苯乙胺高效制备的主要因素,虽然目前关于AmDH的分子改造已有报道,但主要集中在拓展酶的底物谱[22,23],鲜见关于提高苯乙酮催化效率的报道.
本文在源于Bacilluscereus的亮氨酸脱氢酶(BcLeuDH)的催化活性中心引入双突变K70S/N263L,制备了对苯乙酮还原反应具有催化活性的AmDH出发菌株BcAmDH,并利用高效的迭代饱和突变策略对BcAmDH底物结合口袋附近的氨基酸残基进行分子改造,通过高通量筛选获得了对苯乙酮还原反应催化效率显著提高的优势突变株,为(R)-α-苯乙胺的工业合成奠定了基础.
1 实验部分
1.1 试剂与仪器
异丙基-β-D-硫代半乳糖苷(IPTG)、 三羟甲基氨基甲烷(Tris)及咪唑购自上海生工生物工程技术有限公司; 卡那霉素和还原型烟酰胺腺嘌呤二核苷酸(NADH)购于上海索莱宝生物科技有限公司; 苯乙酮、 (R)-α-苯乙胺及(S)-α-苯乙胺购自上海麦克林生化科技有限公司; 其它试剂购自国药集团化学试剂有限公司; 所用试剂均为分析纯.引物合成和质粒测序由无锡天霖生物科技有限公司完成.胺脱氢酶(AmDH)重组表达菌株EscherichiacoliBL21(DE3)/pET-28a-amdh由本实验室构建并保藏.
C1000 Touch型基因扩增(PCR)仪,美国Bio-Rad公司; VCX750型超声破碎仪,美国Sonic公司; Qpix420型高通量菌落挑选仪,美国Molecular Devices公司; Cytation 3型酶标仪,美国BioTek公司; 7890B型气相色谱仪,美国Agilent公司.
1.2 实验过程
1.2.1 同源建模和分子对接 选择源于Geobacillusstearothermophilus的亮氨酸脱氢酶GcLeuDH(PDB登录号: 6ACF; 分辨率0.3 nm; 序列一致性81.3%)的晶体结构为模板[24],运用Swiss-Model服务器(http://swissmodel.expasy.org)建立了AmDH的三维模型,并用Structure Assessment工具对所建模型进行评估[25].从PubChem网站(https://pubchem.ncbi.nlm.nih.gov/)获得苯乙酮的三维结构,用AutoDock软件进行分子对接,用PyMOL软件选择最佳的对接结果并分析复合物的三维模型.通过HotSpot服务器(http://loschmidt.chemi.muni.cz/hotspotwizard/)预测BcAmDH的“热点”氨基酸[26],利用CASTp工具(https://omictools.com/castp-tool)测量酶底物结合口袋的体积[27].
1.2.2 饱和突变文库的构建和筛选 设计含有简并密码子NNK(N=A/T/C/G,K=G/T)的引物,以重组质粒pET-28a-amdh为模板,利用全质粒聚合酶链式反应(PCR)方法(PCR反应条件: 98 ℃变性60 s; 98 ℃预变性30 s,55 ℃退火15 s,72 ℃延伸7 min,30个循环; 72 ℃充分延伸5 min; 16 ℃保存)进行饱和突变以构建突变文库.挑取转化子接种到含有200 μL Luria-Bertani(LB)液体培养基的96浅孔板中,于37 ℃,200 r/min条件下培养8 h后,转接至含有1 mL LB液体培养基的96深孔板中,继续于37 ℃,200 r/min条件下培养3 h至OD600为0.6~0.8; 向培养基中添加终浓度为0.2 mmol/L的IPTG,于20 ℃,200 r/min条件下诱导培养12 h; 向转接后的96浅孔板中加入终体积分数为15%的甘油,于-80 ℃下保藏备用; 将96深孔板于4 ℃,4500 r/min转速下离心10 min收集菌体,并用质量分数为0.9%的生理盐水洗涤2次,向96深孔板中加入500 μL含有终浓度为5 mmol/L苯乙酮的NH4Cl/NH4OH缓冲液(pH=9.5,1 mol/L)重悬浮菌体,于30 ℃,200 r/min条件下反应2 h; 反应液于4 ℃,4500 r/min转速下离心10 min,取100 μL上层清液置于96孔细胞培养板中,加入50 μL 2,4-二硝基苯肼溶液(5 mmol/L)和50 μL NaOH溶液(10 mol/L)显色30 min,用酶标仪测定显色液在530 nm处的吸光值,选取OD530值低于BcAmDH的转化子进行测序及活力测定.
1.2.3 蛋白纯化 将细胞重悬于缓冲液A(100 mmol/L Tris+150 mmol/L NaCl+20 mmol/L咪唑,pH=7.5)中并进行超声破碎,将破碎液于4 ℃,12000 r/min转速下离心30 min; 上层清液用0.22 μm水系滤膜过滤后,缓慢上样至Ni-Nitrilotriacetic acid(Ni-NTA)亲和层析柱; 加样完毕,先用缓冲液A洗涤,再用缓冲液B(100 mmol/L Tris+150 mmol/L NaCl+500 mmol/L咪唑,pH=7.5)进行梯度洗脱,将收集目的蛋白并进行蛋白凝胶电泳(图S1,见本文支持信息).目的蛋白用超滤管脱盐浓缩,并添加终体积分数为20%的甘油,于-80 ℃下保存备用.
1.2.4 酶活力和动力学参数测定 酶活力测定体系总体积为200 μL,包括NH4Cl/NH4OH(pH=9.5,1 mol/L)、 苯乙酮(20 mmol/L)、 NADH(0.2 mmol/L)及适量纯酶液,检测340 nm处的吸光值变化.酶活力单位(U)定义: 在上述条件下,每分钟催化氧化1 μmol NADH的酶活力; 酶比活力(U/g)定义: 每克酶蛋白所含的酶活力单位数.分别在苯乙酮和NADH终浓度为0.5~50.0 mmol/L和0.01~0.2 mmol/L范围内进行活力测定,利用Origin软件中公式Y=VmaxX/(Km+X)进行非线性拟合[式中:Y(U/g)为酶比活力;X(mmol/L)为底物浓度],计算酶动力学常数(Km,mmol·L-1;kcat,min-1)及催化效率(kcat/Km,L·min-1·mmol-1).
1.2.5 苯乙酮的不对称还原反应 反应体系总体积为2 mL,包括BcAmDH或突变株纯酶液(1 mg/mL)、 葡萄糖脱氢酶(1.2 mg/mL)、 苯乙酮(100~300 mmol/L)、 葡萄糖(120~360 mmol/L)、 NAD+(1 mmol/L)、 二甲基亚砜(体积分数20%)和NH4Cl/NH4OH缓冲液(pH=9.5,1 mol/L).反应体系于30 ℃,200 r/min条件下反应,定时取样用NaOH溶液(200 μL,10 mmol/L)淬灭,用甲基叔丁基醚(1 mL)萃取2次.取1 mL萃取液用气相色谱测定苯乙酮转化率,色谱条件: Grace Econo-Cap EC-WAX+色谱柱(30 m×0.25 mm×0.25 μm),柱温程序: 起始温度90 ℃,以20 ℃/min速率升温至160 ℃,保持2 min; 再以10 ℃/min速率升温至200 ℃,保持2 min.另取1 mL萃取液,加入5 mg 4-二甲氨基吡啶和100 μL乙酸酐,于30 ℃,200 r/min条件下衍生化12 h,加入500 μL水淬灭反应; 利用气相色谱测定不对称还原反应产物的构型及其e.e.值,色谱条件: Agilent J&W CP-Chiralsil-DEX CB色谱柱(25 m×0.32 mm×0.25 μm),柱温程序: 起始温度100 ℃,以5 ℃/min速率升温至200 ℃.
2 结果与讨论
2.1 饱和突变位点的选择
利用定点突变引物(表S1,见本文支持信息)在源于Bacilluscereus的亮氨酸脱氢酶的催化活性中心引入双突变K70S/N263L,制备了对苯乙酮催化比活力为90.83 U/g(20 mmol/L,30 ℃)的AmDH出发菌株BcAmDH.
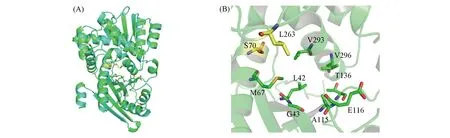
Fig.1 3D structure of GcLeuDH and BcAmDH(A) and distribution of selected amino acid(B) 3D structure of GcLeuDH and BcAmDH are shown as cyan and green cartoon,respectively.The eight selected amino acid residues(L42,G43,M67,A115,E116,T136,V293,V296) and the two residues initially mutated to generate AmDH activity(S70,L263) are shown as green and yellow sticks,respectively.
位于酶底物结合口袋附近的氨基酸突变通常会在一定程度上影响酶的空间结构、 电荷分布及疏水性,从而影响酶的催化活性、 底物专一性及热稳定性等催化特性[28,29].选取与BcAmDH蛋白序列一致性为81.3%的GcLeuDH的晶体结构为模板建立了BcAmDH的三维模型[24][图1(A)],并将底物苯乙酮对接到建立好的三维模型中[25].分析对接结果发现,以苯乙酮为中心0.4 nm范围内有6个氨基酸残基(G43,G44,M67,K82,A115和V293).其中,K82为BcAmDH催化活性中心,G44与底物羰基形成氢键,故选择其余4个残基作为饱和突变的位点.而距离苯乙酮0.4~0.6 nm范围内共有12个氨基酸残基(L42,T45,R46,L63,A81,T83,E116,D117,T136,L263,G292和V296).考虑到此范围内残基个数较多,为了提高筛选效率,进一步对BcAmDH进行基于氨基酸保守性和理化性质分析的“热点”氨基酸预测[26],发现12个氨基酸中有4个(L42,E116,T136和V296)为“热点”氨基酸.综上,共选择L42,G43,M67,A115,E116,T136,V293和V296[图1(B)]8个氨基酸残基进行饱和突变,考察其对苯乙酮还原催化效率的影响.
2.2 正向突变位点的筛选
利用含有简并密码子的引物(表S1,见本文支持信息)进行定点饱和突变以构建突变文库,获得了库容约800株的突变文库.采用基于底物苯乙酮显色反应的高通量筛选方法[30],对所有突变株进行全细胞催化和高通量筛选,在116,136和293位的饱和突变文库中共获得了9株阳性突变株,而在其余5个位点的突变文库中未见阳性突变(图2).

Fig.2 High-throughput screening results of site-directed saturation mutagenesis library(A) and color-rendering results of 116-site saturation mutagenesis library(B) Color-rendering results of BcAmDH and positive mutants are shown in black rectangle and red circle,respectively.
对9株阳性突变株进行了测序分析,结果表明出现6种不同类型的突变.其中,116位出现E116L,E116C和E116V 3种阳性突变类型,136位出现T136G和T136M 2种阳性突变类型,293位仅出现V293A 1种阳性突变类型,所有突变对酶的可溶性表达均未产生明显的不利影响[图3(A)].不同突变株纯酶液对苯乙酮的活性测定结果 [图3(B)] 表明,V293A突变株对苯乙酮的比活力提高幅度最大,与BcAmDH相比提高了124%; T136G和T136M突变株的比活力与BcAmDH相比分别提高了109%和49%; E116L,E116C及E116V突变株的比活力比BcAmDH分别提高了40%,67%和82%.
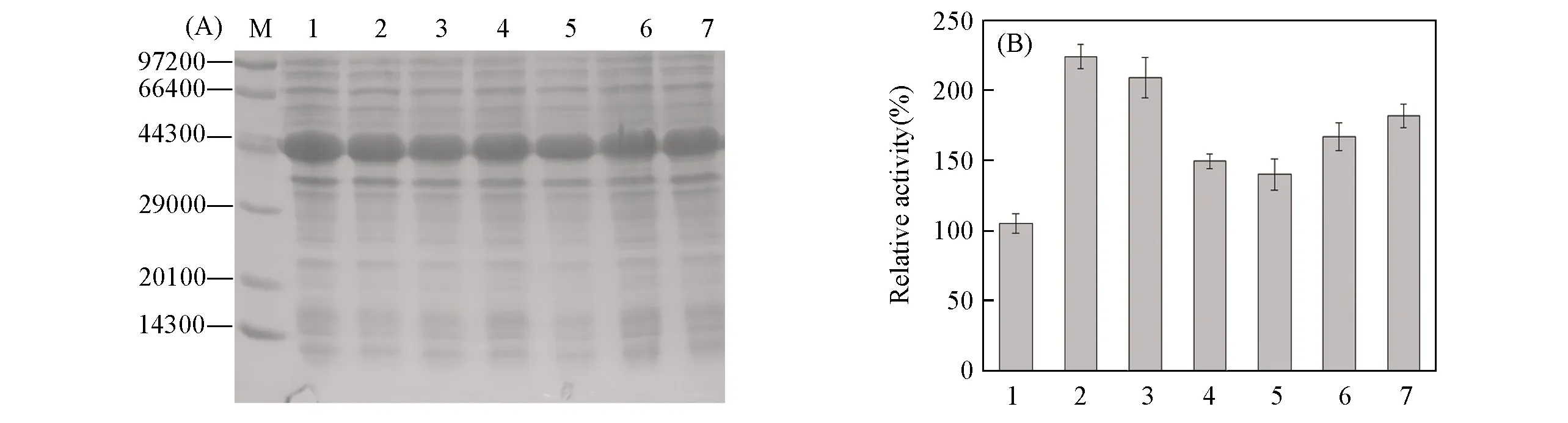
Fig.3 SDS-PAGE analysis(A) and relative activity(B) of positive mutants obtained by site-directed saturation mutagenesis (A) M: low molecular protein marker; lane 1—7 represent crude enzyme for BcAmDH,V293A,T136G,T136M,E116L,E116C and E116V,respectively.(B) 1.BcAmDH; 2.V293A; 3.T136G; 4.T136M; 5.E116L; 6.E116C; 7.E116V.
2.3 迭代饱和突变策略的应用
为了进一步提高酶对苯乙酮的催化活性,选择对3个正向突变位点(116,136和293)进行组合突变.考虑到若将3个位点获得的阳性突变株直接进行组合突变,则可能会遗漏各位点相互作用而产生的新突变类型,同时无法避免其相互作用而产生的负面影响; 若对3个位点进行随机组合突变,当筛选覆盖率达到95%时,需筛选超过90000株转化子,即便选用NDT(N=A/T/C/G,D=A/T/G)密码子,也需筛选大约5200株转化子[31].迭代饱和突变策略是一种按照预先设定的迭代顺序,在每一轮获得的优势突变株的基础上叠加下一轮的饱和突变,经过多次叠加获得性能最优突变株的组合突变方法[32].采用迭代饱和突变对3个位点进行组合突变时,仅需筛选282个转化子即可达到95%的覆盖率[31],可大幅度降低筛选工作量,同时能够有效避免直接组合突变带来的遗漏和负面影响,因此采用迭代饱和突变策略对3个正向突变位点进行组合突变.
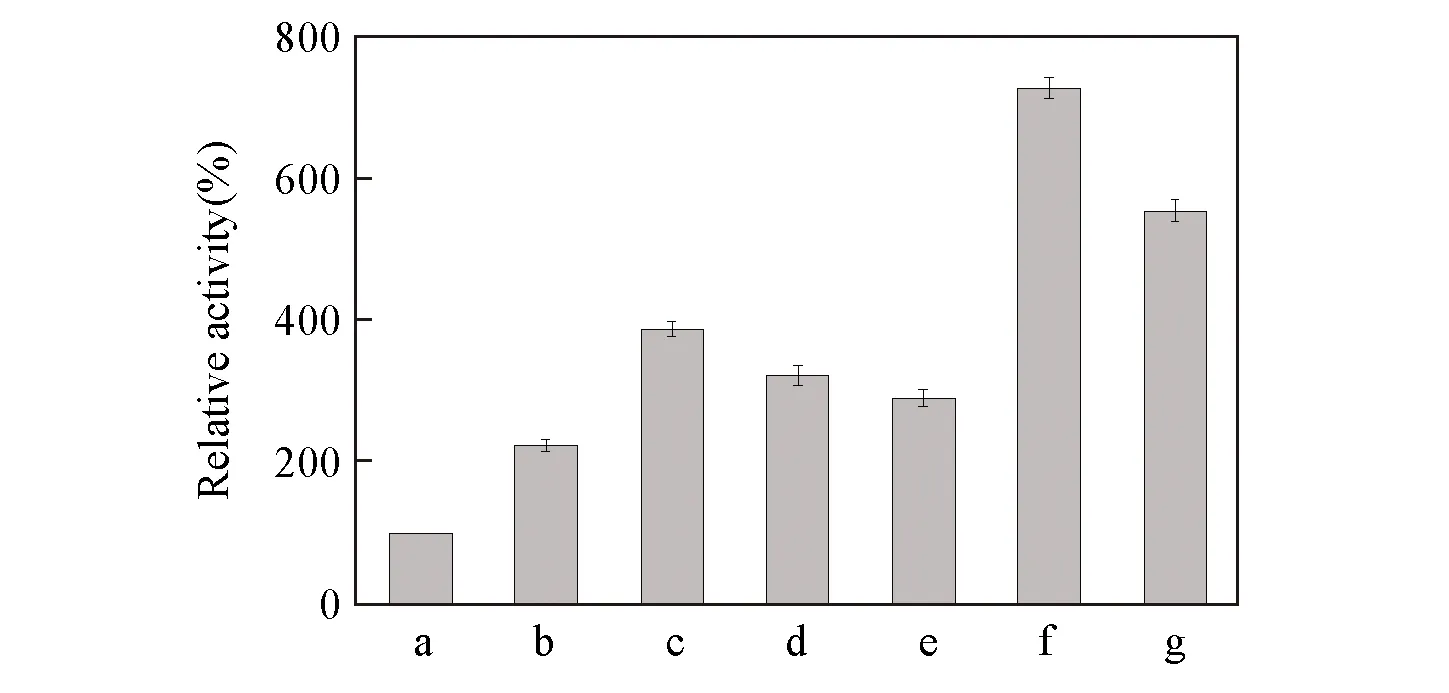
Fig.4 Relative activity of positive mutants obtained by iterative saturation mutagenesis a.BcAmDH; b.V293A; c.V293A/E116V; d.V293A/E116C; e.V293A/E116K; f.V293A/E116V/T136S; g.V293A/E116V/T136C.
基于阳性突变株V293A对苯乙酮的活力提高幅度最大,故选择其作为第一轮迭代饱和突变的模板.由于从116位的饱和突变文库中筛选到多达3个阳性突变株,推测叠加116位的突变后出现阳性突变株的概率较大,首先选择在V293A突变株上叠加116位的饱和突变.经过筛选得到3株阳性突变株V293A/E116V,V293A/E116C和V293A/E116K,其中突变株V293A/E116V对苯乙酮的比活力为352.44 U/g,与BcAmDH相比提高了288%.随后将突变株V293A/E116V作为第二轮迭代饱和突变的模板,叠加136位的饱和突变.经过筛选得到2株阳性突变株V293A/E116V/T136S和V293A/E116V/T136C,最优突变株V293A/E116V/T136S对苯乙酮的比活力达到661.26 U/g,与BcAmDH相比提高了628%(图4).在2轮迭代饱和突变文库中均筛选到了阳性突变株,且出现了定点饱和突变文库中未出现的突变类型(E116K,T136S和T136C),进一步证明了迭代饱和突变策略在组合突变中的实用性和有效性.
测定了BcAmDH及其突变株对底物苯乙酮和辅酶NADH的动力学参数.由表1可见,与BcAmDH相比,V293A突变株对底物苯乙酮的亲和力(Km)略有提高,但同时其转换数(kcat)提高了172%,说明V293A突变株更倾向于提高酶转化底物的速率,从而改善酶的催化效率(kcat/Km).V293A/E116V和V293A/E116V/T136S突变株对底物的亲和力与BcAmDH相当,但在V293A突变株的基础上进一步加快了酶转化底物的速率,从而导致其催化效率与BcAmDH相比分别提高了358%和719%.此外,3株突变株对辅酶NADH的催化效率均有一定程度的改善,与BcAmDH相比分别提高了78%,262%和393%.

Table 1 Kinetic parameters of BcAmDH and three mutants
2.4 关键突变的分析
基于同源建模建立了突变株V293A/E116V/T136S的三维模型[25],并将底物苯乙酮对接到建好的三维模型中[26].比较突变前后的对接结果[图5(A)]发现,底物苯乙酮与突变酶分子之间未形成额外的氢键作用力,但突变酶底物结合口袋附近的空间构型发生了轻微改变.293位的缬氨酸V位于底物结合口袋附近的α-Helix上,且与底物苯乙酮侧链苯环距离较近[图5(B)和 (C)],占据了底物结合口袋的空间,可能会形成空间位阻.当BcAmDH菌株293位的缬氨酸V突变成体积较小的丙氨酸A时[图5(D)],底物结合口袋的体积与BcAmDH相比扩大了0.0224 nm3[27],使得空间位阻减小,提高了其对苯环的接受度,导致酶转化底物的速率加快,V293A突变株对苯乙酮的的催化效率增大.116位谷氨酸E和136位苏氨酸T位于底物进出通道处的Loop上[图5(B)和(E)],通道的构型决定了底物到达酶催化活性中心的难易程度,通常认为比较开阔的通道有利于底物和产物的运输[33,34].当谷氨酸E和苏氨酸T分别突变成缬氨酸V和丝氨酸S后,其侧链长度均有一定程度的减小,导致底物进出通道明显增大[图5(F)],推测E116V和T136S突变使苯乙酮更容易进入到酶催化活性中心,导致酶转化底物的速率进一步加快,V293A/E116V和V293A/E116V/T136S突变株的催化效率逐步提高.
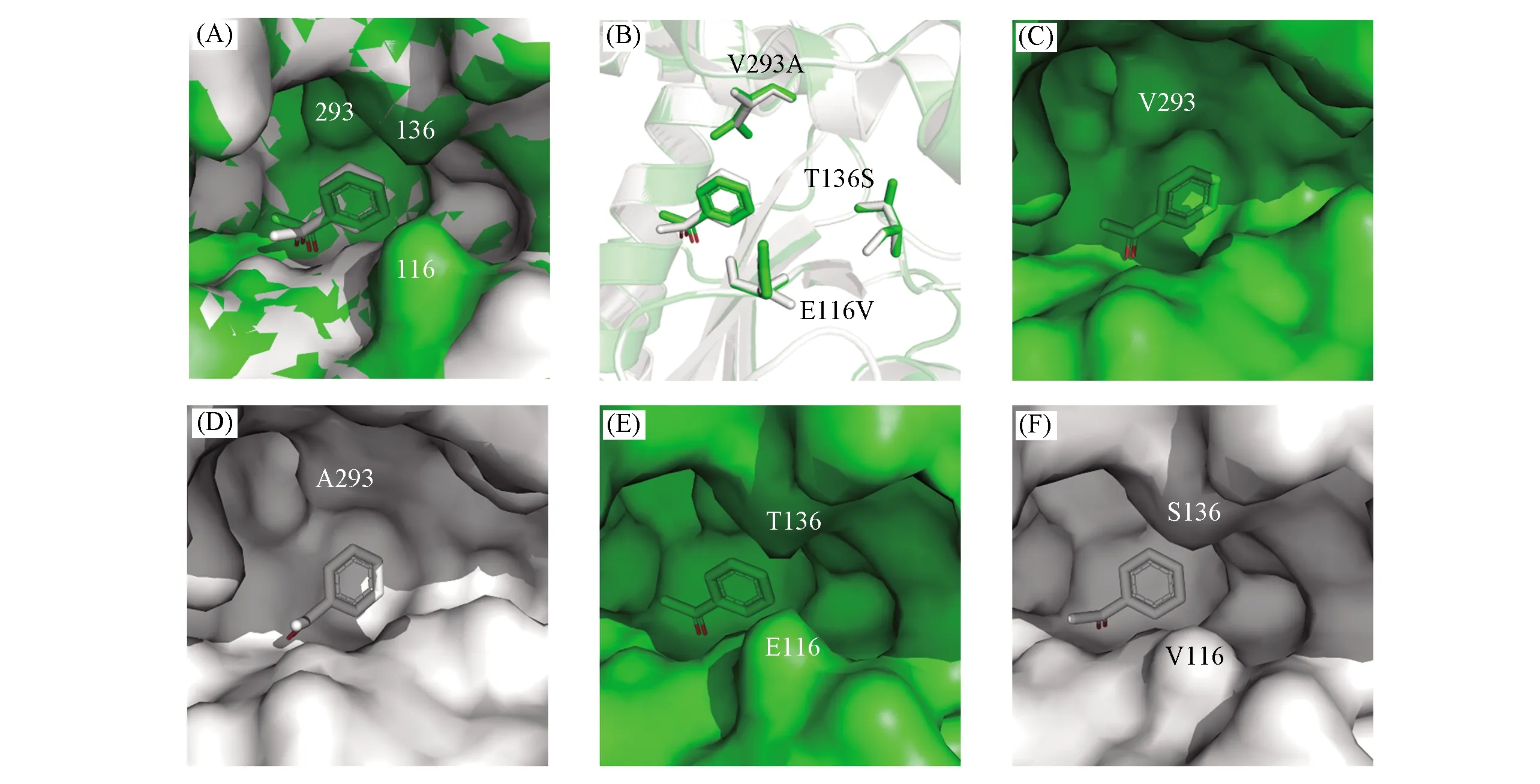
Fig.5 Molecular docking results of BcAmDH and mutant V293A/E116V/T136S with acetophenone (A) Molecular docking results of BcAmDH and mutant V293A/E116V/T136S with acetophenone,3D structures of BcAmDH and mutant V293A/E116V/T136S are shown as green and white surface,116,136 and 293 are the numbering of amino acid residues; (B) molecular docking results of BcAmDH and mutant V293A/E116V/T136S with acetophenone,3D structures of BcAmDHand mutant V293A/E116V/T136S are shown as green and white cartoon,E116V,T136S and V293A refer to mutation results; molecular docking results of BcAmDH(C) and mutant V293A/E116V/T136S with acetophenon(D),showing the effect of V293A mutation near the substrate side chain phenyl group on the shape of the substrate binding pocket; molecular docking results of BcAmDH(E) and mutant V293A/E116V/T136S with acetophenon(F),showing the effect of E116V and T136S mutations at the substrate entry channel on the shape of the substrate binding pocket.The substrate acetophenone is shown as green sticks with BcAmDH and white sticks with mutant V293A/E116V/T136S.
2.5 苯乙酮的不对称还原胺化

Fig.6 Time course of asymmetric reduction of acetophenone with different concentrations by BcAmDH and mutant V293A/E116V/T136S a.100 mmol/L Acetophenone with BcAmDH; b.100 mmol/L acetophenone with V293A/E116V/T136S; c.200 mmol/L acetophenone with V293A/E116V/T136S; d.300 mmol/L acetophenone with V293A/E116V/T136S.
考察了不同底物浓度下BcAmDH和突变株V293A/E116V/T136S作为生物催化剂对不对称还原苯乙酮反应的催化效率.由图6可见,在100 mmol/L底物浓度下,BcAmDH催化反应18 h时苯乙酮转化率为32.1%,48 h时的最终转化率为42.1%; 而突变株V293A/E116V/T136S催化反应18 h时苯乙酮的转化率即达到99.3%,48 h时的最终转化率可达到99.9%,远高于BcAmDH和文献[21]报道值43%(EsAmDH).突变株V293A/E116V/T136S的比转化速率由1.4 mmol/h提高到5.5 mmol/h.当底物苯乙酮浓度增加到200和300 mmol/L时,突变株V293A/E116V/T136S催化反应48 h时的最终转化率分别达到92.3%和80.2%,且不对称还原反应产物(R)-α-苯乙胺的e.e.值为99%(图S2,见本文支持信息),表明突变株V293A/E116V/T136S在较高的底物浓度下仍可以保持高催化效率和优异的立体选择性,显示出其在不对称还原苯乙酮制备(R)-α-苯乙胺过程中的应用潜力.
3 结 论
针对AmDH出发菌株BcAmDH在不对称还原苯乙酮制备(R)-α-苯乙胺过程中酶催化效率低的不足,采用半理性设计对其进行分子改造.利用同源建模法建立了其三维模型并与底物苯乙酮进行分子对接,通过距离原则和“热点”氨基酸预测筛选出8个氨基酸残基进行单点饱和突变,采用基于底物苯乙酮显色的高通量筛选方法获得了3个正向突变位点.进一步采用迭代饱和突变策略对3个正向突变位点进行组合突变,获得的最优突变株V293A/E116V/T136S对苯乙酮还原反应(20 mmol/L,30 ℃)的催化活力和催化效率达到661.26 U/g和2.54 L·min-1·mmol-1,与BcAmDH相比分别提高了628%和719%.使用此突变株催化苯乙酮的不对称还原反应,在100,200和300 mmol/L底物浓度下,转化率分别达到99.9%,92.3%和80.2%,远高于BcAmDH,表明其在不对称合成(R)-α-苯乙胺的过程中具有潜在的应用价值.分子对接结果表明,突变株底物结合口袋的空间位阻减小和底物进出通道的扩大是催化效率提高的主要原因.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20190621.