胶束萃取-浊点预富集-高效液相色谱法同时测定山楂中9种酚酸和黄酮类化合物含量
2020-05-16胡玉周光明罗庆红李幼林
胡玉,周光明,罗庆红,李幼林
胶束萃取-浊点预富集-高效液相色谱法同时测定山楂中9种酚酸和黄酮类化合物含量
胡玉,周光明,罗庆红,李幼林
西南大学化学化工学院发光与实时分析教育部重点实验室,重庆 400715
建立以非离子表面活性剂、胶束萃取-浊点预富集与超声萃取技术结合的HPLC方法,同时测定山楂中原儿茶酸、咖啡酸、牡荆素、阿魏酸、金丝桃苷、槲皮素、木犀草素、山柰酚和芹菜素的含量。以非离子表面活性剂Genapol X-080为萃取剂,加入一定量氯化钠进行浊点预富集,以萃取剂浓度、固液比、电解质浓度、溶液pH值、超声时间、平衡时间、离心时间为考察因素,9种成分的提取总量为考察指标,优化胶束萃取和浊点预富集参数。采用Phenowenex C18色谱柱,甲醇-0.06%冰醋酸溶液为流动相,梯度洗脱,检测波长290 nm。该条件下9种成分的分离度良好,标准曲线线性范围良好,>0.999 0(=8)。加样回收率分别为:原儿茶酸(99.7±1.23)%、咖啡酸(97.5±2.49)%、牡荆素(105.1±1.50)%、阿魏酸(105.5±0.76)%、金丝桃苷(100.5±2.41)%、槲皮素(102.3±1.86)%、木犀草素(107.0±1.52)%、山柰酚(97.7±1.72)%、芹菜素(99.9±2.34)%。本研究建立的方法可完成山楂中9种成分的分离和测定,缩短样品前处理时间,简便快速,环境友好,可为山楂中黄酮和有机酸的提取提供可靠的分析方法。
胶束萃取;浊点预富集;高效液相色谱法;Genapol X-080;山楂
山楂为蔷薇科植物山里红Bge.var.N.E.Br.或山楂Bge.的干燥成熟果实,具有消食健胃、行气散瘀、化浊降脂功效[1]。山楂含有大量黄酮类化合物,如牡荆素、金丝桃苷等,是抗癌作用较强的成分,且山楂所含大量有机酸可帮助消化[2-3],因此,山楂被广泛用于药品和食品中。目前,测定山楂中黄酮和酚酸的含量通常采用HPLC[3-5]、高效薄层色谱法[6]和红外光谱法等,样品前处理通常采用液-液萃取法[7]、索氏萃取法[8]和固相萃取法[9-10],此类方法提取多数情况采用有机溶剂萃取法,存在有机溶剂用量大、处理时间长、温度高而易破坏有效成分、环境污染严重、操作繁琐等缺点。因此,研究山楂中酚酸及黄酮类化合物的高效、低毒、经济的提取方法,对于开发中药新品种具有重要意义。
胶束萃取-浊点预富集以水代替影响环境的有机溶剂作为萃取剂,通过加入少量非离子表面活性剂产生增溶作用和浊点现象,从而实现有效提取[11-12]。表面活性剂由疏水基和亲水基组成,分别与水和待提取的有效成分结合,形成疏水基团在内、亲水基团在外的胶束,从而提高有机物在水中的溶解度[13],即表面活性剂的增溶作用。同时,表面活性剂水溶液加热到浊点温度以上时,溶液变浑浊,静置一段时间后出现分相,当外界条件反向变化时,两相消失,再次浑浊,称为浊点现象[14-15]。由于无机盐的阴阳离子与表面活性剂竞争水分子而导致盐析,以及强化表面活性剂的水化而导致盐溶,且氯化钠的溶解度受温度影响较小,故试验中加入氯化钠可降低非离子表面活性剂的浊点,使浊点萃取在较低温度下发生。选择表面活性剂时,需考虑其对待测样品和实际操作的影响,以及浊点萃取法作为预处理步骤时表面活性剂对检测仪器的影响,如在紫外区是否有强的吸收而干扰样品的测定。本试验采用胶束萃取技术分离目标分析物,应用HPLC检测其含量[16-17],以期为药食两用中药山楂的质量控制提供参考依据。
1 仪器与试药
LC-20A高效液相色谱仪(包括SPD-20A紫外检测器、CTO-10AS柱温箱、2个LC-20AD泵),日本岛津公司;KQ3200型超声波清洗器,昆山市超声仪器有限公司;SZ-2自动双重纯化水蒸馏器,上海泸西分析仪器厂有限公司;FA2004A型分析天平,上海精天电子仪器有限公司;SHZ-D循环水式多用真空泵,重庆东悦仪器有限公司;80-1(Ⅲ)电动离心机,金坛市科析仪器有限公司。
山楂样品购自蒙山九品堂旗舰店,经鉴定为蔷薇科植物山楂Bge.的干燥成熟果实;对照品原儿茶酸(批号Z30M6L1)、咖啡酸(批号27649)、牡荆素(批号J1212059)、阿魏酸(批号E1420043)、金丝桃苷(批号MUST-13061701)、槲皮素(批号M02118066)、木犀草素(批号36414)、山柰酚(批号A1306003)和芹菜素(批号K1404029),上海晶纯实业有限公司,纯度均大于98%;Genapol X-080(批号9043-30-5),苏州亚科科技股份有限公司;甲醇(色谱纯)、冰醋酸(分析纯),重庆川东化工有限公司化学试剂厂;二次蒸馏水,本实验室自制。
2 方法与结果
2.1 溶液制备与试验条件
2.1.1 对照品溶液制备
精密称取原儿茶酸、咖啡酸、牡荆素、阿魏酸、金丝桃苷、槲皮素、木犀草素、山柰酚、芹菜素对照品,分别移至10 mL量瓶中,用甲醇溶解,稀释并定容,摇匀,制得浓度分别为560、820、840、630、530、450、610、400、700 μg/mL的对照品溶液。
用移液管精密量取原儿茶酸、槲皮素各3 mL,金丝桃苷、山柰酚各4 mL,其余对照品溶液各1 mL于10 mL量瓶中,用甲醇稀释至刻度,得到混合对照品溶液,置4 ℃冰箱中避光保存,有效期60 d。
2.1.2 样品处理
①胶束萃取:准确称量0.1 g干燥山楂样品粉末,量取4 g/100 mL Genapol X-080水溶液10 mL,置15 mL离心管中,磁力搅拌混合均匀,放入50 ℃水浴,超声(功率500 W,频率60 kHz)萃取40 min,3500 r/min离心15 min,取上清液,0.45 μm有机滤膜过滤,待用[18-19]。②浊点预富集:将上述萃取上清液转入10 mL离心管中,加入氯化钠1.46 g,旋涡振荡仪振荡5 min,置入50 ℃恒温水浴中平衡40 min,3500 r/min离心15 min,使两相分离,弃去下层水相,分取上层表面活性剂富集相,用甲醇定容至2.0 mL以降低表面活性剂黏度,经0.45 μm有机滤膜过滤,供HPLC分析。
2.1.3 色谱条件
色谱柱:Phenomenex C18柱(150 mm×4.6 mm,5 μm);流动相:A为0.06%乙酸水溶液,B为甲醇;线性梯度洗脱:0~5 min,40%→65%B;5~10 min,65%→70%B;10~15 min,70%→90%B;15~20 min,90%→40%B;流速:0.8 mL/min;进样量:20 μL;紫外检测波长:290 nm;柱温:35 ℃。
2.1.4 富集效果评价
采用富集因子(enrichment factor,EF)表征对目标分析物的富集效果,以浊点预富集后的溶液浓度(C1)与预富集前的溶液浓度(C0)比值计算,即EF=C1/C0。
2.2 流动相选择
分别考察乙腈-水、乙醇-水、甲醇-水为流动相体系对分离效果的影响,结果表明,以乙醇-水为流动相时色谱峰有重叠,以乙腈-水和甲醇-水分别作为流动相时均能分离,从经济和环保角度考虑[20],选择甲醇-水为流动相。为避免组分峰形不对称,造成前展和拖尾现象,故在流动相水中加入少量醋酸。本试验采取梯度洗脱,在色谱柱允许的pH值范围内加入不同比例乙酸于水溶液中,最后确定“2.1.3”项下的最佳流动相和洗脱条件。
2.3 不同萃取剂比较
在相同的色谱条件下,分别研究100%甲醇、50%甲醇、乙醇、丙酮4种常用萃取剂与4 g/100 mL Genapol X-080对目标分析物提取量的影响,结果表明,采用非离子表面活性剂Genapol X-080为萃取剂时,获得的目标分析物总提取量最高。
2.4 胶束萃取-浊点预富集参数优化单因素试验
2.4.1 萃取剂浓度
考察Genapol X-080用量对目标分析物萃取效率的影响,由于表面活性剂黏度较大,取量时有误差,将其稀释为20 g/100 mL。萃取剂浓度分别为1、2、3、4、6、8 g/100 mL对提取量的影响见图1。可以看出,浓度为4 g/100 mL时萃取效率最高。分析原因为萃取剂浓度较低时分析物难以完全进入有机相,浓度过高则降低了有机相目标分析物的浓度。
2.4.2 固液比及波长
在波长250~340 nm范围内检测混合对照品和样品,结果波长<270 nm时黄酮类成分的紫外吸收弱,波长>330nm时有机酸的紫外吸收弱,故选择290 nm为检测波长,9种目标分析物均有较强的吸收峰。比较不同固液比对目标分析物提取量的影响,将山楂样品质量固定为0.1 g,改变萃取剂溶液体积(分别为2、4、6、8、10、12 mL),结果见图2。可以看出,体积为2~10 mL时提取量逐渐增加,体积>10 mL时提取量基本不变,表明体积为10 mL时超声萃取基本达到平衡,再增加样品溶液体积会稀释目标成分浓度,无法提高EF[16],因此,选择10 mL作为样品溶液体积。

图1 萃取剂浓度对山楂中9种成分提取量的影响

图2 固液比对山楂中9种成分提取量的影响
2.4.3 溶液pH值
目标分析物含有羧基和羟基,所以在水中存在的分子态和离子态随溶液pH值的改变而改变,不同的存在状态影响其在有机相和水相的分配系数,最终影响富集效果。考虑目标分析物的稳定性,以纯冰醋酸溶液调节溶液的pH值,分别加入0、0.1、0.2、0.3、0.4、0.5 mL冰醋酸,对提取量的影响见图3。可以看出,随溶液酸性增强,EF逐渐增大。选择0.4 mL冰醋酸调节溶液pH值为5.6。
2.4.4 电解质种类及浓度
考察3种电解质(氯化钠、碳酸钠、硫酸钠)对萃取剂富集效果的影响,结果表明,加入氯化钠可以使样品提取量明显提高,因此选择氯化钠电解质增强离子强度。同时考察了不同浓度(1.0、1.5、2.0、2.5、3.0、4.0 mol/L)电解质对目标分析物萃取效率的影响,结果见图4。当氯化钠浓度为2.5 mol/L时,萃取效率最大,因此最终选择电解质浓度为2.5 mol/L。
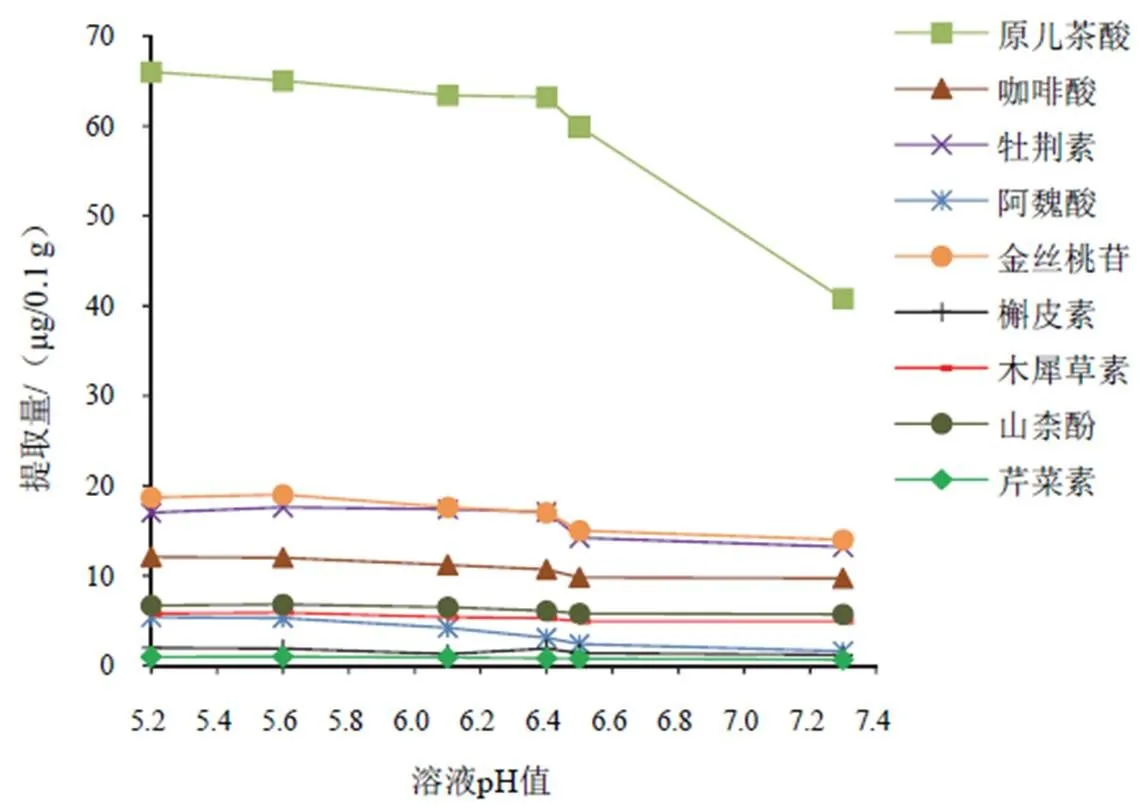
图3 溶液pH值对山楂中9种成分提取量的影响

图4 电解质浓度对山楂中9种成分提取量的影响
2.4.5 超声时间
超声处理可使萃取剂与样品溶液充分接触和混合,样品溶液在表面活性剂作用下形成乳浊液,萃取剂与水相之间接触面积增大,有利于目标分析物由水相转入萃取剂中。采用体积为10 mL的样品溶液,超声功率为500 W,考察不同超声时间(10、20、30、40、50、60 min)对目标分析物提取量的影响,结果见图5。可以看出,随着超声时间延长,目标分析物提取量逐渐增加,30 min后,提取量有所下降。由于超声会产生热量,超声时间过长可能增加目标分析物成分挥发程度和解离程度[21],因此选择超声时间为30 min。
2.4.6 平衡时间
经过超声的两相溶液加入电解质氯化钠,在水浴中静置一定时间,使目标分析物从水相移至有机相中,最终达到平衡。考察平衡时间10、20、30、40、50、60 min对提取量的影响,结果见图6。可以看出,平衡时间为30 min时萃取效果最好,延长时间则萃取效果不变,故选择30 min为平衡时间。
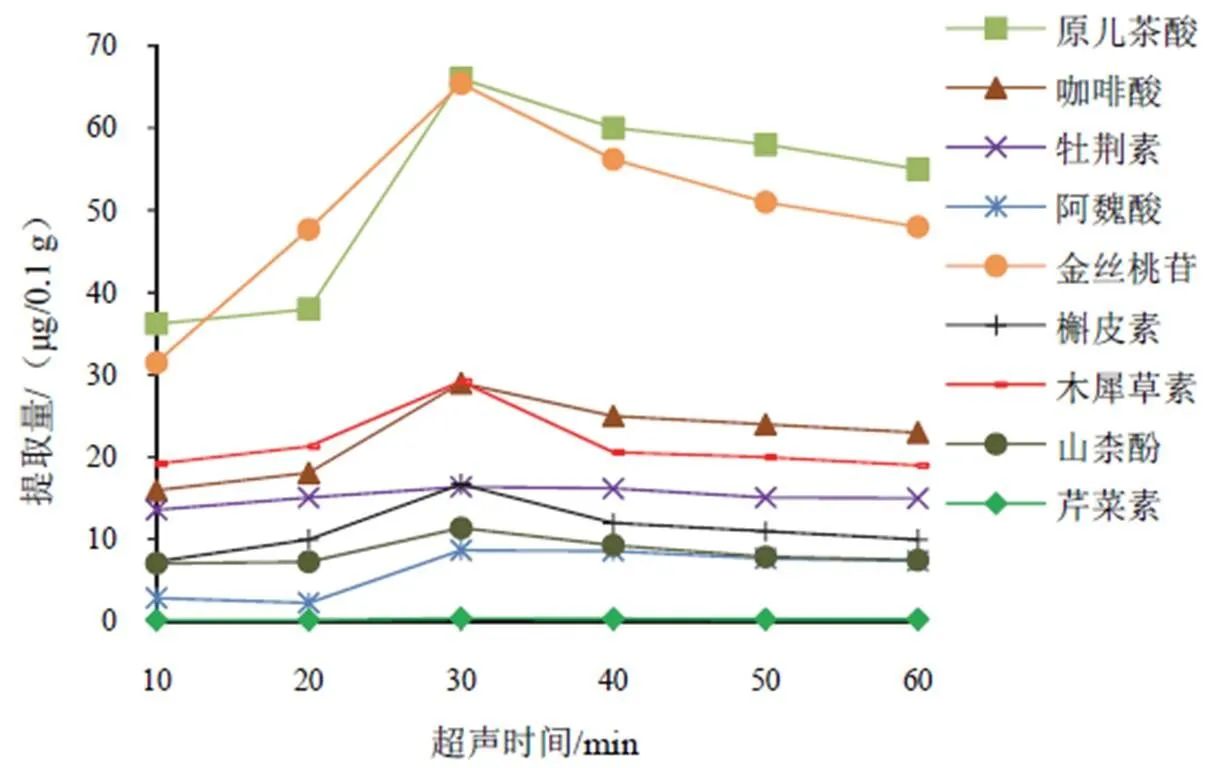
图5 超声时间对山楂中9种成分提取量的影响

图6 平衡时间对山楂中9种成分提取量的影响
2.4.7 离心时间
离心可使分散在水溶液中的萃取剂聚集,由于表面活性剂密度较大,在溶液下层,有利于萃取剂与水相的分离。考察离心时间为5、10、15、20、25、30 min对提取量的影响,结果见图7。可以看出,离心时间为25 min时提取量最高。离心时间过短则表面活性剂未完全聚集,离心时间过长则温度升高,表面活性剂熔化而发生损失[22],因此选择25 min为离心时间。

图7 离心时间对山楂中9种成分提取量的影响
2.5 胶束萃取-浊点预富集参数优化正交试验
在单因素试验基础上,选择萃取剂浓度、固液比、溶液pH值、电解质浓度、超声时间、平衡时间、离心时间7个因素分别进行2个水平试验,采用L8(27)正交试验,以9种活性成分的提取总量评价提取性能,进一步优化各参数。正交试验设计结果及直观分析见表1。从极差R可知,对提取总量影响最大的因素是萃取剂浓度和固液比,其次是溶液pH值和萃取时间,平衡时间、离心时间和电解质浓度的影响很小,与单因素试验结果有一定差异,可能是多因素间相互影响所致。从直观分析结果可知,最佳提取工艺条件为:固液比1∶100(g/mL),萃取剂浓度4 g/100 mL,超声时间30 min,平衡时间30 min,离心时间25 min,电解质浓度2.5 mol/L,溶液pH值5.6。
表1 山楂中9种成分提取工艺正交试验设计结果与直观分析
试验号萃取剂浓度/(g/100 mL)固液比/(g/mL)超声时间/min平衡时间/min离心时间/min电解质浓度/(mol/L)溶液pH值提取总量/(μg/0.1 g) 13 83020252.55.6265.0 23 83030303.05.2263.8 34 84020253.05.2276.9 44 84030302.55.6289.3 53104020302.55.2275.0 63104030253.05.6287.6 74103020303.05.6291.9 84103030252.55.2282.5 K11091.410951103.21108.811121111.81133.8 K21140.611371128.81123.311201120.21098.2 R 49.2 42 25.6 14.4 8 8.4 35.6
2.6 方法学考察
2.6.1 专属性试验
取空白4 g/100 mL Genapol X-080水溶液10 mL,按“2.1.3”项下色谱条件,进样20 μL,色谱图见图8A;按“2.1.1”项下方法制备混合对照品溶液,按“2.1.3”项下色谱条件,进样20 μL,色谱图见图8B。可以看出,表面活性剂水溶液的紫外吸收很弱,不干扰目标分析物的检测。预富集前后样品溶液色谱图见图8C、图8D,可以看出,预富集后的溶液中活性成分含量高于预富集前。

注:A.空白Genapol X-080水溶液;B.混合对照品液;C.预富集前样品溶液;D.预富集后样品溶液;1.原儿茶酸;2.咖啡酸;3.牡荆素;4.阿魏酸;5.金丝桃苷;6.槲皮素;7.木犀草素;8.山柰酚;9.芹菜素
2.6.2 线性关系考察
取“2.1.1”项下混合对照品溶液,配制成8个不同浓度梯度的混合对照品溶液,按“2.1.3”项下色谱条件,从低到高浓度分别进样20 μL,进行检测。以峰面积为纵坐标,对照品浓度为横坐标,进行线性拟合,得到回归方程。山楂中9种成分的回归方程、线性范围、相关系数和检出限(信噪比为3)见表2。根据检出限,表明对照品在相应的线性范围内与峰面积成良好的线性关系,该方法有较高的灵敏度和较高的检测范围。
表2 9种成分线性关系考察结果
成分回归方程线性范围/(μg/mL)相关系数检出限/(μg/mL) 原儿茶酸Y=-9475.1+40 749.1X0.000 119~42.00.999 60.000 036 咖啡酸Y=3596.8+64 233.7X0.000 155~20.50.999 50.000 047 牡荆素Y=-1969.1+81 915.6X0.000 112~21.00.999 00.000 034 阿魏酸Y=-10 138.6+140 039.6X0.000 257~15.80.999 50.000 078 金丝桃苷Y=-1516.9+35 341.5X0.000 099~53.00.999 00.000 030 槲皮素Y=55 322.8+61 211.4X0.000 092~33.80.999 80.000 028 木犀草素Y=17 447.2+59 535.1X0.000 221~30.50.999 70.000 067 山柰酚Y=-18 267.9+51 953.7X0.000 211~40.00.999 30.000 064 芹菜素Y=28 550.8+89 574.4X0.000 178~17.50.999 70.000 054
2.7 精密度、重复性及稳定性试验
移取混合对照品溶液,在上述色谱条件下连续进样6次,根据相应对照品的峰面积,计算得到9种成分RSD为1.25%~1.34%,表明仪器精密度良好。精密称取同一批山楂样品粉末5份,按“2.1.2”项下方法制备样品溶液并检测,结果9种成分峰面积RSD为0.97%~1.99%,表明该方法重复性良好。移取同一样品溶液,于0、6、12、16、20、24 h分别进行检测,结果显示9种成分峰面积RSD为0.98%~2.14%,表明该方法具有良好的稳定性。
2.8 加样回收率试验
精密称取已知含量的同一批干燥山楂样品粉末各0.1 g,每3份为一组,按低、中、高浓度分别加入相应对照品溶液,按“2.1.2”项下方法制备供试品溶液,分别进样测定,计算加样回收率,结果见表3。
表3 9种成分加样回收率试验
成分取样量/g样品含量/μg加入量/μg测得量/μg回收率/%平均回收率/%RSD /% 原儿茶酸0.1074.160134.3100.1 0.1176.275149.2 98.7 99.71.23 0.1075.380155.4100.0 咖啡酸0.1029.725 53.2 97.3 0.1131.330 58.8 95.9 97.52.49 0.1130.135 64.7 99.4 牡荆素0.1017.015 33.8105.6 0.1018.418 38.7106.4105.11.50 0.1218.220 39.5103.3 阿魏酸0.10 9.4 8 18.3105.4 0.10 9.310 20.5106.3105.50.76 0.1110.112 23.1104.7 金丝桃苷0.1174.260132.2 98.5 0.1073.872145.5 99.8100.52.41 0.1075.480160.3103.2 槲皮素0.1014.310 25.3104.1 0.1113.214 27.3100.3102.31.86 0.1013.715 29.4102.4 木犀草素0.1117.814 33.5105.2 0.1117.317 36.9107.6107.01.52 0.1016.419 38.3108.3 山柰酚0.1011.810 21.6 99.3 0.1212.112 23.1 95.9 97.71.72 0.1112.815 27.2 97.9 芹菜素0.10 1.610 11.4 97.9 0.11 1.212 13.1 99.4 99.92.34 0.10 1.315 16.7102.5
2.9 样品含量测定
取山楂样品粉末各0.1 g,按“2.1.2”项下方法制备3份样品溶液,每份溶液进样3次测定分析,根据线性方程计算样品中各指标成分含量,结果见表4。
表4 山楂样品中各成分含量测定(μg)
成分123平均值 原儿茶酸75.976.576.276.2 咖啡酸31.231.431.331.3 牡荆素18.018.618.618.4 阿魏酸 8.7 9.5 9.7 9.3 金丝桃苷72.774.274.573.8 槲皮素13.113.513.013.2 木犀草素17.316.817.817.3 山柰酚12.012.611.612.1 芹菜素 1.4 1.3 0.9 1.2
3 讨论
本试验采用超声辅助胶束萃取-浊点预富集法,通过单因素试验和正交试验确定了最佳提取条件:表面活性剂浓度4 g/100 mL,固液比1∶100(g/mL),超声时间30 min,平衡时间30 min,离心时间25 min,电解质浓度2.5 mol/L,溶液pH值5.6。其中对提取结果影响最大的是萃取剂浓度和固液比。
本试验通过高效液相色谱法实现了对山楂中9种活性成分同时分离和定量检测,在以表面活性剂作萃取剂的基础上,添加浊点预富集步骤,可以提高有机酸和黄酮的提取量。结果表明,山楂中9种成分的含量为:原儿茶酸76.2 μg/0.1 g、咖啡酸31.3 μg/0.1 g、牡荆素18.4 μg/0.1 g、阿魏酸9.3 μg/0.1 g、金丝桃苷73.8 μg/0.1 g、槲皮素13.2 μg/0.1 g、木犀草素17.3 μg/0.1 g、山柰酚12.1 μg/0.1 g、芹菜素1.2 μg/0.1 g。加样回收率试验结果表明,该方法稳定性强,检测限低,可为山楂和其他中药有机酸和黄酮类化合物的提取和研究提供可靠的参考依据。
[1] 国家药典委员会.中华人民共和国药典:一部[M].北京:中国医药科技出版社,2010:87.
[2] 时岩鹏,丁杏苞.山楂化学成分的研究[J].中草药,2000,31(3):173-174.
[3] 王超群,郭敏,易敏之,等.山楂化学成分及高效液相色谱分析方法研究进展[J].中国中医药信息杂志,2006,13(11):93-95.
[4] 李博艺,谌柄旭,魏志阳,等.高效液相色谱法测定山楂黄酮的研究[J].中国酿造,2018,37(3):157-161.
[5] 王珍,李秀霞,李娇,等.高效液相色谱法测定山楂中芦丁、金丝桃苷和槲皮素的含量[J].食品工业科技,2013,34(9):302-305.
[6] SRIVASTAVA N, SRIVASTAVA S, VERMA S, et al. Evaluation of quantitative variation of secondary metabolites in(Haw.) using high performance thin layer chromatography[J]. The Journal of Biomedical Research,2014,28(4):328-331.
[7] 曹学丽,杨春蕾.基于液-液萃取机理的新型环境样品前处理方法研究进展[J].中国农学通报,2011,27(6):242-248.
[8] 冯尚彩,王爱香,任吉同.山楂叶中金丝桃苷的提取与测定方法研究[J].临沂大学学报,2010,32(6):65-68.
[9] 李桂珍,唐为扬,曹伟敏.印迹分子聚合物结合低共熔溶剂用于山楂中咖啡酸的固相萃取[J].色谱,2015,33(8):792-798.
[10] ZHOU Y, KONG W, LI Y, et al. A new solid-phase extraction and HPLC method for determination of patulin in apple products and hawthorn juice in China[J]. Journal of Separation Science,2012, 35(5/6):641-649.
[11] 申进朝,邵学广.浊点萃取技术及其在有机化合物分离分析中的应用[J].化学进展,2006,18(4):482-487.
[12] 刘超,罗晶,马辉,等.浊点萃取法提取贯叶连翘总黄酮的研究[J].西北药学杂志,2012,27(4):296-298.
[13] 王志龙.非离子表面活性剂为溶媒的浊点萃取技术[J].化学工程, 2006,34(4):9-12,19.
[14] ZHOU J, WANG S W, SUN L X. Determination of ostholeinrat plasma by high-performance liquid chromatography using cloud-point extraction[J]. Anal Chim Acta,2008,608(2):158.
[15] SAGARADZE V A, BABAEVA E Y, KALENIKOVA E I. HPLC-UV method for determing flavonoids in hawthorn flowers and leaves[J]. Pharmaceutical Chemistry Journal,2017,51(6):277-280.
[16] 杨晓博,王荣芳,贾亚楠,等.高效液相色谱法测定山楂叶中的9种酚类成分[J].食品工业科技,2017,38(10):62-66.
[17] 谢振伟,但德忠,赵燕,等.超声波辅助萃取技术在样品预处理中的应用[J].化学通报(网络版),2005,68(1):1-11.
[18] 卢彦芳,安静,孙婷,等.微波辅助胶束萃取-高效液相色谱法同时测定平消片中马钱子碱、士的宁和柚皮苷的含量[J].北京中医药大学学报, 2011,34(2):126-130.
[19] 姜兰芳,周光明,李艳艳.胶束萃取-高效液相色谱法同时测定葛根粉中5种异黄酮[J].食品科学,2011,32(6):186-190.
[20] CHANG Q, ZHU M, ZUO Z, et al. High-performance liquid chromatographic method for simultaneous determination of hawthorn active components in rat plasma[J]. Journal of chromatography B, Biomedical Sciences and Applications,2001, 760(2):227-235.
[21] YANG J Z, WEI W, ZHU W X, et al. High performance liquid chromatography tandem mass spectrometry method for determination of patulin in apple, hawthorn and tomato products[J]. Food Science,2009,30(4):162-165.
[22] WANG S Y, CHAI J Y, ZHANG W J, et al. HPLC Determination of five polyphenols in rat plasma after intravenous administrating hawthorn leaves extract and its application to pharmacokinetic study[J]. Yakugaku Zasshi,2010,130(11):1603-1613.
Simultaneous Determination of Nine Phenolic Acids and Flavonoid Compounds in Crataegi Fructus by Micelle Extraction-Cloud Point Preconcentration-HPLC
HU Yu, ZHOU Guangming, LUO Qinghong, LI Youlin
To establish an HPLC method combining non-ionic surfactant with micelle extraction- cloud point preconcentration and ultrasonic extraction technology; To simultaneously determine the contents of 9 active components in Crataegi Fructus, including protocatechuic acid, caffeic acid, vitexin, ferulic acid, hyperin, quercetin, luteolin, kaempferol and apigenin.Non-ionic surfactant Genapol X-080 was used as the extraction agent, and a certain amount of sodium chloride was added to perform cloud point preconcentration. With total contents of 9 active components as index, factors such as extraction agent concentration, solid liquid volume, electrolyte concentration, pH value of solution extraction, ultrasonic time, equilibrium time and centrifugal time were investigated to optimize the parameters of micellar extraction and cloud point preconcentration. Phenowenex C18 chromatographic column was used, with methanol-volume fraction 0.06% glacial acetic acid solution as mobile phase and gradient elution, and detection wavelength was 290 nm.The separation of 9 active components was completely achieved under the condition. Calibration curves of the nine components showed good linear relationship (>0.999 0,=8). The average recovery rates were protocatechuic acid (99.7±1.23)%, caffeic acid (97.5±2.49)%, vitexin (105.1±1.50)%, and ferulic acid (105.5±0.76)%, hyperin (100.5±2.41)%, quercetin (102.3±1.86)%, luteolin (107.0±1.52)%, kaempferol (97.7±1.72)%, and apigenin (99.9±2.34)%.The method established in this research can complete the separation and determination of 9 active components in Crataegi Fructus, shortening the sample pretreatment time. The method is simple, rapid and environment friendly, which can provide a reliable analytical method of extraction of flavonoids and organic acids in Crataegi Fructus.
micelle extraction; cloud point preconcentration; HPLC; Genapol X-080; Crataegi Fructus

R284.1
A
1005-5304(2020)05-0058-07
10.3969/j.issn.1005-5304.201905375
国家自然科学基金(21675131);中央高校基本科研业务费专项(100030-2120130993)
周光明,E-mail:gmzhou@swu.edu.cn
(2019-05-27)
(2019-06-20;编辑:陈静)
