广东省主要红树林植物DNA条形码评价1)
2020-05-13武锋裴男才廖宝文管伟姜仲茂李玫
武锋 裴男才 廖宝文 管伟 姜仲茂 李玫
(中国林业科学研究院热带林业研究所,广州,510520)
2003年Hebert et al.[1]第一次提出识别物种的新方法——DNA条形码技术。两年后,该技术的相关理论被引入植物学开展科学研究[2-3]。随后国际生命条形码联盟植物工作组(Consortium for the Barcode of Life Plant Working Group)初步确定并推荐叶绿体基因片段的rbcL和matK[4]作为的DNA核心条形码片段。然而该片段并不能像动物的COI片段一样具有普遍适用性。因此叶绿体基因非编码区trnH-psbA[5-6]及核基因ITS[7-9]作为核心条形码的补充片段被提了出来。选择、比较、推荐合适的DNA条形码片段的工作也一直在继续[10-13]。DNA条形码技术不仅能应用于相似生物的识别[14-17]。还可以利用DNA条形码片段测序所获得的分子序列,构建特定生物类群的系统发育关系[18-20]。这将促进群落生态学、系统发育生物学、生态学与条形码技术的交叉融合,带来进化生物学等相关学科的整合与发展[21-25]。
红树林是生长在热带、亚热带海陆过渡地区的木本植物群落[26],具有重要的生态服务价值,在世界16种主要生态系统中排名第四[27]。全世界共有红树植物84种[28]。广东共有红树林植物25种[29],占世界红树林物种数的29.76%。相对以往对红树林的研究主要集中在宏观方面,如红树林生态系统服务功能评价、碳汇、防风消浪等,近年来对红树林DNA条形码[30]、基因组[31-32]等微观分子方面的研究也越来越多。然而对红树林的准确识别是开展各项研究的基础。利用红树林植物的外部形态来不能快速、准确的区分相同属相似物种,比如海桑属(Sonneratia)的杯萼海桑(SonneratiaalbaSm.)、海桑(Sonneratiacaseolaris(L.) Engl.)、海南海桑(SonneratiaxhainanensisW. C. Ko et al.)、拟海桑(SonneratiaparacaseolarisW. C. Ko et al.)等;木榄属(Bruguiera)的海莲(Bruguiera.sexangula(Lour.) Poir)、尖瓣海莲(Bruguiera.sexangulavar.rhynchopetalaKo.)等。传统的植物群落发育关系构建中,如APG分类系统(Angiosperm Phylogeny Group),多是以大类群植物间的系统发育关系,对大多数植物只能解决到目或科的水平,不宜于科及以下分类水平的研究,进化树分辨率低[28]。同时运用该方法得出的系统发育树容易出现多分枝结构。DNA条形码以动物[33]、热带、亚热带植物[21,34]、微生物[35]为对象的研究在物种识别和系统发育关系构建方面都取得了一定的成果。Saddhe et al.[30]利用核心条形码片段和ITS2片段对印度西海岸5科14种红树林的识别也取得了成功,但是该研究对象与分布在中国的红树林植物多有不同,同时该研究的对象仅包含真红树,植物种类少。红树植物群落物种丰富度较低,类群间关系比较简单。是否利用DNA条形码序列也能成功构建红树植物系统发育关系答案尚不明确。本文尝试研究DNA条形码在中国红树林(包含真红树和半红树)植物类群中的通用性,并构建红树林植物系统发育关系,为红树林生物多样性保护提供科学依据。
1 材料与方法
本研究选取红树林在广东的主要分布地,统筹考虑选取珠三角地区的深圳、惠州,粤东的汕头和粤西的湛江等4地市的红树林分布区(珠海淇澳岛作为补充采样点),具体的采样地信息详见表1。根据Li等[9]和高连明等[36]DNA条形码样品采集规范,每个红树林物种采样2~3个个体,主要以采取新鲜叶子、新芽为主,便于提取DNA分子材料,每个个体相距20 m以上。采集完试验材料立刻用硅胶干燥。共采集红树林植物144个个体。经廖宝文研究员、李玫副研究员鉴定,确定类属于16科22属23种(其中2种属于红树林伴生种),全部正确分类到种。本研究采样的物种数占广东红树林植物真红树和半红树种类25种的84%,因其中人工引种的海莲和尖瓣海莲几乎灭绝、消失,故未采样;小花老鼠簕(AcanthusebracteatusVahl)、钝叶臭黄荆(PremnaobtusifoliaR. Br.)未找到。
1.1 DNA提取、扩增和测序
采用调整过的CTAB法[37]提取红树植物DNA。根据国际生命条形码联盟推荐以及前人[6,38-39]在区域性植物DNA条形码的研究,选取叶绿体基因片段rbcL、matK以及trnH-psbA总计3个分子序列作为研究的扩增片段。引物信息和扩增程序参考表2。所有扩增产物经凝胶电泳检测后送广州美吉生物公司完成测序,3种片段均采取双向测序,获得的序列在https://www.ncbi.nlm.nih.gov/上进行BLAST搜索,如发现序列与采样物种存在科、属不一致的情况,咨询专家再次确认或重新取样扩增,直到所有序列的BLAST结果与采集样品为同属或者同科植物。使用DNAstar中的SeqMan(5.0.0)对测序获得的序列进行拼接和校对。然后在Geneious11.1.3中选择MAFFT Aligement插件,按默认参数进行序列比对。
注:A代表湛江;B代表深圳;C代表惠州;D代表汕头;E代表珠海。
1.2 数据分析
参考Kress et al.[40]计算PCR扩增成功率和测序成功率方法。PCR扩增成功率是指某片段扩增的成功个体数占该片段所有个体的百分数。测序成功率是指获得的高质量序列数占所有个体的百分数。研究中采用了序列相似法(Basic Local Alignment Search Tool,BLAST)和系统发育树法中的邻接树(Neighbor-Joining tree,NJ tree)法来评估红树林植物种的鉴定能力。(1)BLAST方法,首先在Geneious 11.1.3中将3种DNA片段分别建立一个本地数据库[40],将所有序列比对后另存为*fasta文件,调整序列方向,清除序列间gap。再用blast+2.7.1+把每条序列与数据库内的所有序列进行BLAST,以相同位点(Identical Sites)的百分比作为量化标准。若同物种的Identical Sites最小值都大于与其他所有物种个体间的Identical Sites值,那么我们认为这个物种的序列得到了准确鉴定,若该物种只包含一个样品,为避免过高估计鉴定成功率,标记为鉴定失败[41]。鉴定成功率是通过成功鉴定物种数的百分比再乘以该片段的测序成功率得到的。联合片段的鉴定成功率与单个片段计算方法相同。(2)NJ tree方法,用rbcL、matK、trnH-psbA3个单片段或者片段组合,在MEGA6中利用基于Kimra’2-parameter model的NJ法构建系统发育树,并完成1000次运算获得节点支持率。当同一物种的所有个体聚为一枝,并且节点支持率≥70%,则认为鉴定成功[39,42]。所有单个个体的物种被用来构建NJ树,但是在分辨率计算时不被计算在内[43]。
从NCBI网站下载小花老鼠簕、钝叶臭黄荆、海莲的rbcL、matK和trnH-psbA片段信息各1条(无尖瓣海莲相关条形码片段信息),与采集到的红树林植物的序列信息拼接,组成一个包含26种物种,49个个体的序列文件。用于构建红树林植物系统发育树。
2 结果与分析
2.1 序列通用性
对23种红树林植物的144个个体进行序列统计,共获得3种DNA条形码片段可用序列386条(表3),序列获得率89.35%。其中,在湛江红树林保护区共收集红树植物16种,35个个体,测序后共得到89条序列。PCR扩增后发现rbcL和trnH-psbA片段的扩增成功率同为最高100%,其次是matK71.43%。测序成功率最高为rbcL片段100%,trnH-psbA片段次之88.57%,最低为matK片段65.71%。
在深圳红树林保护区共收集红树植物12种,36个个体,测序手得到99条序列。PCR扩增后3片段的扩增成功率由高到低依次为rbcL和trnH-psbA100%、matK(80.56%)。测序成功率最高为rbcL片段(100%),trnH-psbA片段次之(97.22%),最低为matK片段(77.78%)。
在惠州红树林保护区共收集红树植物14种,40个个体,测序后得到108条序列。3个片段扩增成功率由高到低依次为rbcL片段100%、trnH-psbA97.5%,matK77.5%。测序成功率最高为rbcL片段100%,trnH-psbA次之92.5%,最低为matK77.5%。
在汕头红树林保护区采集到红树植物9种,24个个体,测序后共得到67条序列。PCR扩增后3片段的扩增成功率由高到低依次为rbcL、trnH-psbA(100%)、matK(91.67%)。测序成功率最高为rbcL和trnH-psbA片段100%,matK片段为79.17%。
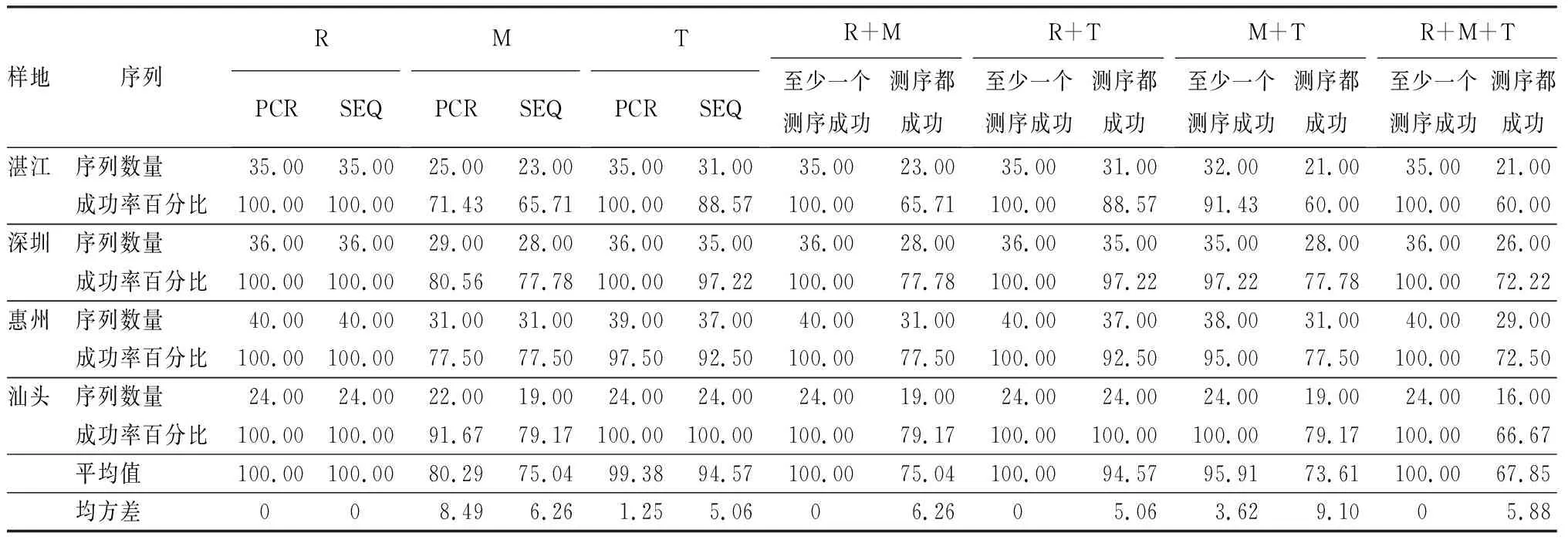
表3 广东省红树林植物DNA片段扩增和测序成功率 %
注:珠海作为补充采样点,共采集植物3种9个样品,获得序列23条,用于后续构建系统发育树,此处未做统计。PCR(Polymerase Chain Reaction):多聚酶链式反应;SEQ(Sequence):测序。R为rbcL;M为matK;T为trnH-psbA。
2.2 物种鉴定成功率
2.2.1 序列相似性分析
因分布在广东的红树、半红树植物除海桑属和老鼠簕属外均是单属、单种存在,故这里仅统计红树植物的种的鉴定成功率。BLAST结果显示,单个片段中对物种的鉴别成功率最高为trnH-psbA84.48%±12.09%,其次依次为rbcL片段82.16%±9.68%,最低为matK片段65.09±6.00%,trnH-psbA片段和rbcL片段的物种识别率显著高于matK片段(表4)。然而除汕头地区,rbcL片段成功鉴定的物种均高于trnH-psbA片段,主要差别为汕头地区红树林中trnH-psbA能准确区分开无瓣海桑和海桑,而rbcL不行。
对多片段的不同组合统计分析,发现核心条形码rbcL+matK的鉴定成功率最高,为84.58%±10.83%;rbcL+trnH-psbA鉴定成功率次之,为83.71%±15.96%。补充条形码trnH-psbA联合rbcL+matK片段后物种鉴定率反而降低到76.89%±8.07%。
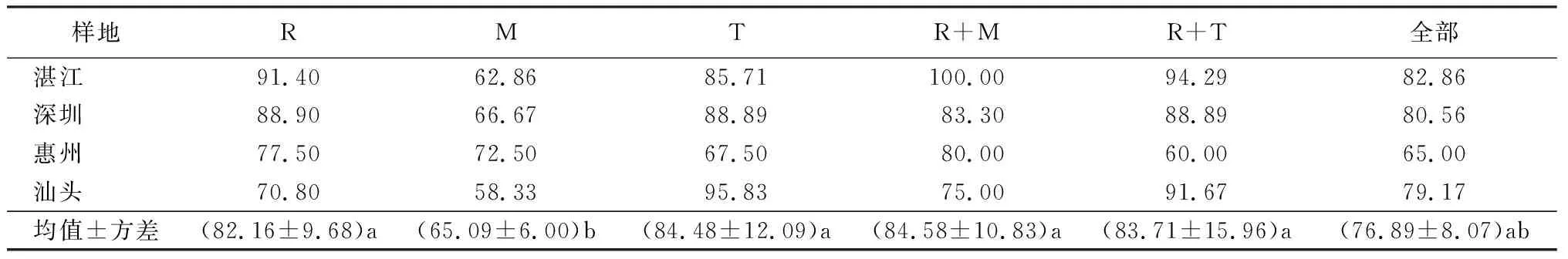
表4 利用序列相似法的单片段和多片段DNA条形码在红树林种上的鉴别成功率 %
注:R为rbcL;M为matK;T为trnH-psbA。
2.2.2 系统发育分析
运用系统发育方法分析统计物种识别率结果如表5。结果显示rbcL物种识别率最高,为66.65%±17.35%;其次是trnH-psbA,为61.21%±9.34%,matK最低为47.75%±13.23%。片段联合时rbcL+trnH-psbA的物种识别率为62.23%±9.47%;rbcL+matK62.54%±17.41%,再增加trnH-psbA片段可以提高到65.15%±11.09%。
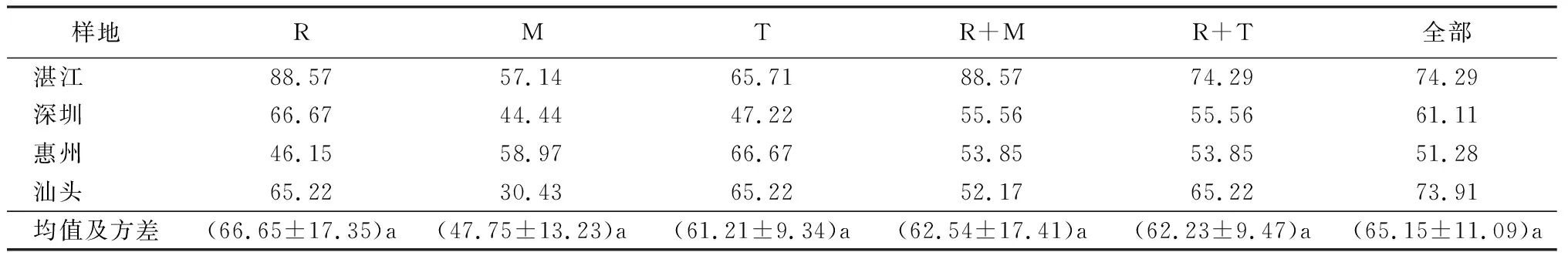
表5 利用系统发育法的单片段和多片段DNA条形码在红树林种上的鉴别成功率 %
注:R为rbcL;M为matK;T为trnH-psbA。
利用rbcL、matK和trnH-psbA单个或3个片段的联合片段,采用NJ法构建系统发育树(图1)。结果发现,利用rbcL单一片段即可构建平均节点支持率最高的红树林植物系统发育树。除老鼠簕外,同一物种首先聚为一支,同属物种再聚为一支(绿色标记),然后同科的物种关系最近(黄色标记)。
3 结论与讨论
本文尝试利用DNA条形码技术对广东省红树林植物进行研究,目的在于评估DNA条形码在红树林植物种的引物通用性、鉴别成功率、系统发育树构建等方面的表现。
3.1 DNA条形码在红树林群落中的通用性
在本研究中,核心条形码中的rbcL片段在红树植物DNA样品中的PCR扩增和测序成功率都达到了100%,获得序列条带信息、质量良好。与Kress et al.[21]研究相比、表现出更高的通用性和成功率。与裴男才[44]研究同处于热带、亚热带的森林植物群落的测序成功率相比(90%-100%)持平。这也与Saddhe et al.[45]研究印度西海岸红树植物的扩增和测序成功率(97.7%)不相上下。说明通用片段rbcL可变位点最少,所选引物序列通用性强。推荐其成为红树林DNA条形码片段。而另一核心片段matK的PCR扩增成功率和测序成功率均是3个片段中的最低水平,其中利用该片段对角果木、秋茄扩增或测序不成功,在木榄、卤蕨、红海榄等物种只有少量个体测序成功,说明该片段引用的通用性不强,可能是由于存在单核苷酸重复序列的原因。如Costion et al.[46]研究中matK片段的扩增率也只有68.18%;Parmentier et al.[47]的研究中,matK片段的扩增成功率仅为64%。刘娟[39]、卢孟孟等[42]、魏亚男等[41]的研究中也存在类似情况,普遍修正做法是增加引物数量进行多次尝试。而Saddhe et al.[45]的研究中,matK片段的扩增成功率达到95.5%,这主要是因为其采用的引物不同。同时印度西海岸的红树植物与中国广东分布的红树植物种类不同也是造成扩增成功率差异较大的原因。在以后的研究中可尝试利用其研究中采用的matK引物。补充条形码trnH-psbA的引物通用性好,仅用一对引物即可使样品的扩增率和测序成功率保持在88%以上,仅次于rbcL片段,同时该片段能成功区分海桑属植物。综上所述,推荐rbcL、trnH-psbA作为红树林DNA条形码的研究片段。
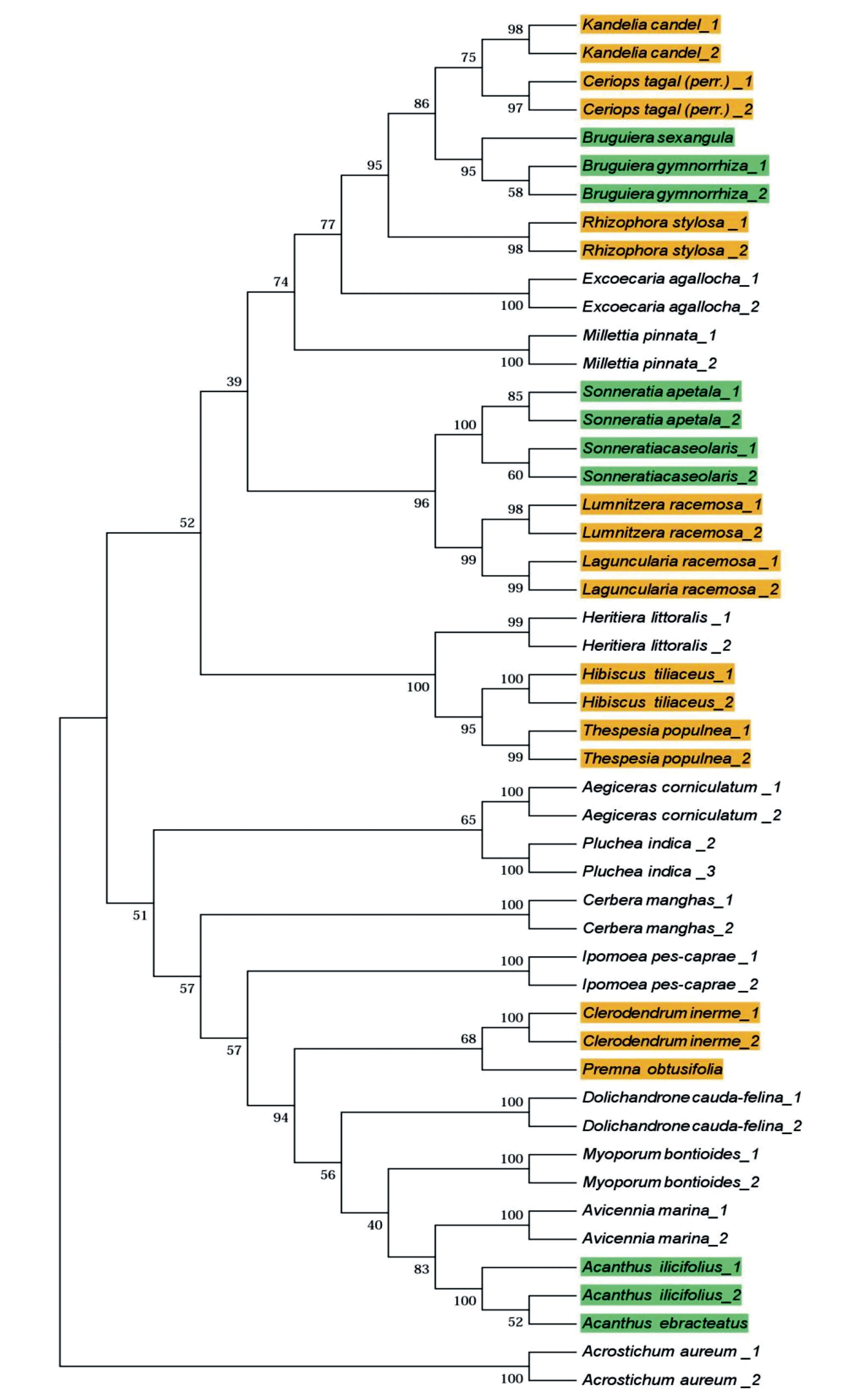
图1 运用rbcL片段构建的广东省红树林植物群落系统发育树
3.2 物种鉴定能力
本文采用两种不同的方法来评价DNA条形码对红树林植物的识别能力。获得的结果也有不同。比如利用BLAST方法物种识别率最高的单片段是trnH-psbA;利用NJ方法物种识别率最高的片段是rbcL。且同一片段或者片段组合的物种识别率均是BLAST法大于NJ树法,且差距较大。这与卢孟孟等[42]的研究结论一致。主要因为前者仅比较位点之间的差异,后者还要兼顾所有个体是否形成单系,同时隐存种的存在也会降低NJ树获得的物种识别率。此外相对于前人要求节点支持率大于50%[41,47]或60%[48],本文要求节点支持率大于70%,也是NJ法物种识别率降低的一个原因。同时多片段的组合分析还会降低物种识别率,如利用BLAST法rbcL+matK片段物种识别率为84.58%±10.83%,rbcL+matK+trnH-psbA后为76.89%±8.07%;NJ树法中,单片段rbcL物种识别率为66.65%±17.35%,rbcL+trnH-psbA后为62.23%±9.47%。主要是因为trnH-psbA片段变异较大,存在多个插入或者缺失的碱基,造成同物种个体间Identical Site值变小,导致物种识别率下降。
利用BLAST法分析发现matK片段物种的识别率为65.09%±6.00%,这与前人进行植物DNA条形码的研究结果大体一致[38,48]。尽管trnH-psbA在不同植物类群间碱基数目差异很大,存在大量的插入和缺失造成在红海榄、木榄、阔苞菊等物种中测序困难。但在本研究中trnH-psbA的测序成功率与rbcL无显著差异,都明显高于matK片段。同时该片段能独立使用时成功鉴别海桑属的海桑和无瓣海桑。Gonzalez et al.[49]等和Tripathi et al.[50]研究也表明trnH-psbA是最具希望和潜力的条形码之一。
NJ法分析显示同一物种聚为一枝,4个地区存在大量相似的物种,但不同地区间相似物种的DNA条形码片段物种识别率率无显著差异。相似物种的存在可能反映了红树林物种起源和系统发育框架的相似性。同时由于红树林地理位置的不同,所处的环境条件也不同。这些差异可能导致4个地区的红树林物种向不同方演变。由南到北,真红树植物种类逐步减少。
Burgess et al.[40]在加拿大温带植物群的研究中,发现核心条码组合可以成功鉴定93%的物种,以及De Vere et al.[43]发现rbcL+matK可以鉴别威尔士境内69.4%~74.1%的有花植物。Kress et al.[21]对Panama区域内296个木本物种进行研究,发现叶绿体片段rbcL+matK的物种识别率高达98%。本研究中核心条码rbcL+matK组合片段物种平均识别率也可以达到84.58%±10.83%,与rbcL+trnH-psbA片段组合的物种平均识别率无显著差异。然而无论采用哪种分析方法,片段组合rbcL+matK的物种平均识别率与单片段rbcL的相比无显著提高。说明rbcL是片段组合rbcL+matK中物种识别的主要片段。综上所述,本文推荐rbcL、trnH-psbA作为红树林植物DNA条形码片段。
3.3 系统发育树
热带的巴罗科罗拉多岛(Barro Colorado Island,BCI)[21]、海南热带云杉林[51],亚热带的鼎湖山样地[44],哀劳山森林[42]等地利用rbcL+matK+trnH-psbA片段组合构建系统发育树均获得成功。然而在对处于相同纬度带红树林植物群落构建系统发育树时却未能成功。rbcL+matK片段组合也存在类似情况。
只用单一片段rbcL可成功构建系统发育树,且发育树的节点平均支持率也最高,该片段在Saddhe et al.[45]研究的印度西海岸的真红树群落中也获得成功。即使加上从网站下载的红树林植物DNA条形码片段信息后,该片段对应的物种也能成功归类,这也印证了系统发育树法下rbcL片段的物种识别率最高。此外,我们发现各分枝上节点的平均支持率均大于50%,说明红树林树种的进化关系具有较高的可靠性。同时利用DNA条形码获得的系统发育树框架末端都获得了很好的二岐分枝结构,系统发育树末端分枝为分类单元提供了准确的系统位置,可根据DNA条形码片段来计算整个群落内所有类群内部真实的分枝长度。相比于传统的系统发育树建树方法,利用DNA条形码获得的系统发育树在进化树的分辨率和进化时间的准确性方面都更具优势。Sahu et al.[52]用rbcL、matK和ITS片段基于最大似然法(Maximun likelihood,ML)成功构建印度东、西部海岸37种红树林植物和63种非红树植物组成的系统发育树。相比于最大似然法,邻接树法运算更快,可以满足构建小群落的系统发育树。故推荐用rbcL片段基于邻接树法构建广东红树林植物系统发育树。
总的来说,rbcL和trnH-psbA片段扩增成功率、测序成功率高,说明这两种条码在红树树种中是通用的。在物种识别方面,3种片段中rbcL和trnH-psbA的物种识别率相对较高,片段组合未能明显提高红树植物物种识别率。同时仅用一对引物的rbcL片段即可准确构建出广东省红树林植物群落系统发育关系。trnH-psbA片段作为叶绿体基因非编码区序列,进化速率快,且能准确区分某些rbcL不能识别的物种,可以作为补充片段。综合比较,rbcL、trnH-psbA是值得推荐为红树林植物DNA条形码片段。
本研究得到23种红树林植物3种DNA条形码片段386条有效序列,红树林植物种类占中国真红树和半红树的55.26%。后续可继续扩大采样范围,补充其他红树林树种及红树林伴生植物的DNA条形码,构建一个完整、高覆盖度的红树林植物DNA条形码数据库。
