Triplet Excited States in Organic Luminescent Materials
2020-04-26XiaoLeiXinZhangChunFeng
Xiao Lei-Xin, Zhang Chun-Feng
National Laboratory of Solid State Microstructures, School of Physics,and Collaborative Innovation Center for Advanced Microstructures, Nanjing University, Nanjing, 210093
Abstract: Organic luminescent materials are promising for next-generation flexible optoelectronics.Nonetheless, the luminescent efficiency in organic molecules is limited by a barrier between the singlet and triplet excited states.In this mini review, we focus on the strategies to overcome such limit by manipulating the dynamics of triplet excited states.By designing the electronic coupling between singlet and triplet excited states, the processes of hot intersystem crossing, reversal intersystem crossing and triplet state stabilization at different stages are successfully implemented to promote the light emission in the organic luminescent materials.The understanding of the mechanisms drives the development of thermally-activated delayed fluorescence and organic long-persistent luminescence,which are promising for the applications of organic light-emitting diodes, sensors and bioimaging.
Key words: organic luminescent material; triplet excited state; charge transfer state; charge separated state;hot exciton;hot intersystem crossing;reverse intersystem crossing;triplet state stabilization; thermally activated delayed fluorescence; long persistent luminescence; organic light-emitting diode

Organic luminescent materials have already gained worldwide market share of light-emitting devices(LEDs)[1−17].Benefiting for the mechanical flexibility and cost effectiveness, organic luminescent materials are expected for potential applications beyond organic LEDs including lasers, sensors and bioimaging[18−22].Different from most inorganic semiconductor light emitters, spin multiplicity is particularly important in organic molecules.The gap between the singlet and triplet excited states is much larger than the strength of spin-orbital coupling in most molecules without heavy atoms.Thus, the intersystem crossing(ISC) is generally inefficient in comparison with other competing channels (Figure 1).Consequently, the quantum efficiency of fluorescence-based LEDs is limited to below 25%.Technically, efficient organic LEDs are demonstrated using phosphorescence induced by recombination from triplet excited states[22].
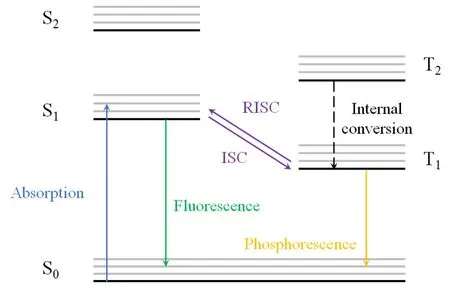
FIG.1.Diagram for fundamental transition processes taking place in organic system after photoexcitation.The lowest singlet excited states (S1) are generated by absorbing photo energy.The S1 states can undergo the intersystem crossing (ISC) process to be converted to the lowest triplet excited states (T1) and vice versa.Transitions between states of the same spin multiplicity are called internal conversion(IC).Fluorescence and phosphorescence are resulted from the radiative transitions from the S1 and T1 state to the ground state.
The carrier dynamics for light emission from conventional luminescent molecules are summarized in Figure 1.With different spin configurations, the excited states are catalogued as singlet and triplet states.The internal conversion process between two singlet or triplet excited states, regulated by Kasha’s rule, are generally much faster than the ISC process.Fluorescence emission from singlet excited states (S1) to the ground state is electrical dipole allowed.The radiative recombination process is competed against by nonradiative pathways, setting the typical lifetime for fluorescence emission on the scale of 1-100 ns.Phosphorescence emission exhibits a much longer lifetime due to the low transition possibility from the lowest triplet excited states(T1)to the singlet ground states(S0).Nevertheless,the efficiency of slow phosphorescence recombination may be significantly reduced by the thermallyactivated non-radiative recombination channels[20].As an example, the phosphorescence from the aromatic molecules can only be observed under low temperatures.
The rate of ISC is dependent on the spin-orbital coupling(⟨S|ˆHSOC|T⟩)and the energy difference(∆ST)between singlet and triplet states[21].In many organic luminescent materials of aromatic carbonyl compounds, the exited state is the admixture ofn →π∗and π→π∗configurations[22].The ISC rate is relatively large if the transition between singlet and triplet states involves a change of molecular orbital type, known as El-Sayed’s rule[23].The coupling between singlet and triplet states with the same molecular orbital types is usually weak.In this case, the process of ISC becomes efficient only when the energy difference(∆ST)is small and comparable to the hyperfine interaction[24].
These principles have recently been examined in several sophisticated systems.Some fascinating effects have been demonstrated with promising potential applications.In a few systems,the ISC process is possibly enhanced by the strong coupling between highly excited singlet and triplet states[25−29].The hot ISC process may be sufficient to compete with the pathway of internal conversion,mixing the singlet and triplet excited states for highly efficient luminescence.In multiple systems with the energy difference ∆STcomparable to the thermal fluctuation, the reversal ISC process at room temperature may become an efficient channel to convert triplet population to singlet states[19,22].Such a thermally-activated delayed fluorescence (TADF) process may promote the quantum efficiency of fluorescence emission beyond 25% in electroluminescence devices.Moreover, in the condensed phases of organic molecules, molecular aggregation may stabilize triplet excited states of different molecule orbitals.The stabilized triplet states may contribute to luminescent emission of extremely long lifetime, known as organic long persistent luminescence (OLPL)[22,30−32].These processes dramatically modify the transitional opinion and stimulate promising applications.In this mini review,we focus on understanding the critical role played by triplet excited states in the hot ISC, TADF and OLPL processes in a variety of organic molecules.
I.HOT ISC PROCESS
Excitons located at the upper excited state, which is higher than the lowest excited state, are called hot excitons[26].Fluorescent materials that can convert triplet hot excitons into singlet excitons open up a new pathway for harvesting the non-emissive triplet excitons[26−29].The relatively shorter lifetime of the triplet hot excitons in these fluorescent materials significantly reduces the possibility of the accumulation of triplet excitons and as a result suppresses the triplettriplet annihilation (TTA) and singlet-triplet annihilation (STA) process[22].Therefore, OLEDs based on“hot excitons” materials usually exhibit a small efficiency roll-off when the current increases.The reversal ISC process from the triplet hot excitons to the singlet excitons was firstly observed by Keller in 1969[33],based on the weak fluorescence emission observed in several aromatic molecules.In 2014, a highly efficient near-infrared (NIR) OLED based on the “hot exciton”material was developed, exhibiting an external quantum efficiency (EQE) of 1.54% together with a small efficiency roll-off[26].
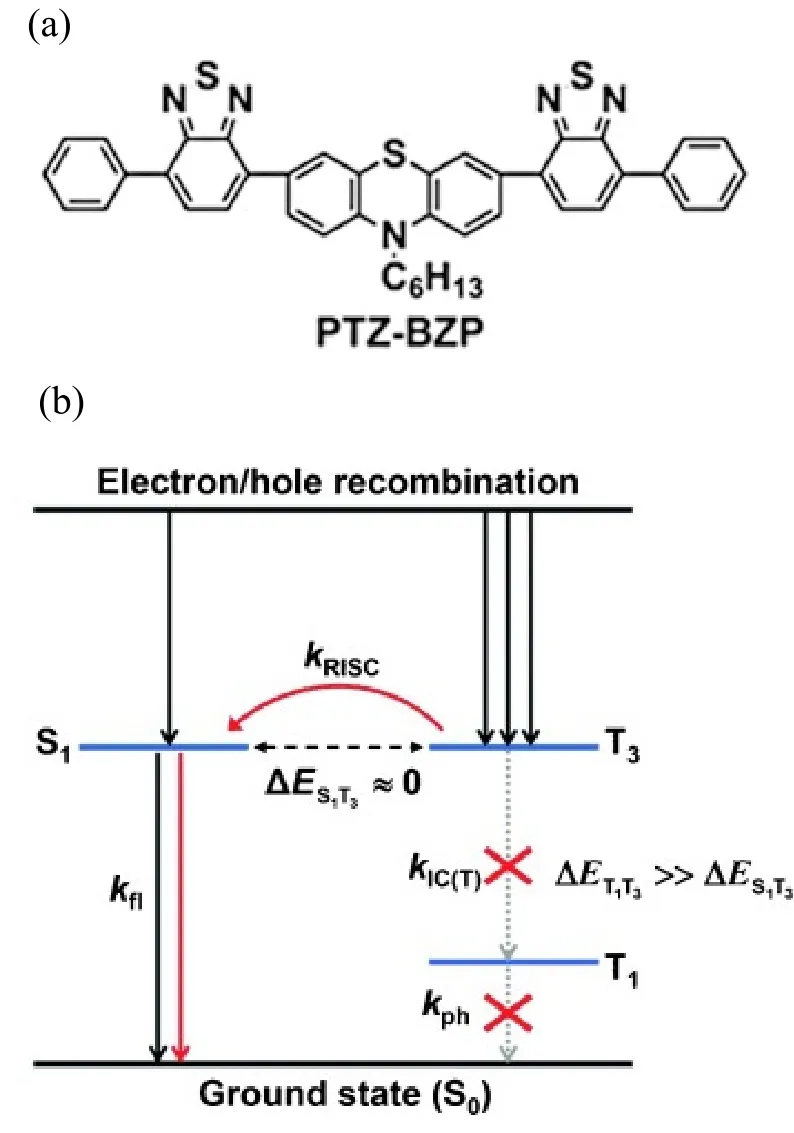
FIG.2.(a) The molecular structure of PTZ-BZP.(b) The model for exciton relaxation in the EL process.The large energy gap between the T3 and T1 states reduces the rate of internal conversion.The RISC process from the T3 state to the S1 state is efficient due to the small energy gap between them.Reproduced with permission from ref.26.Copyright 2014 John Wiley & Sons.
According to the Kasha’s rule,electron occupied at a high-lying excited state usually relaxes to the lowest excited state very fast in most organic molecular systems[29].In some materials,a large energy gap between the high-lying T2and T1states suppresses the internal conversion from the T2to T1states, resulting a relatively long lifetime of hot triplet excited states[26−29].Such a hot ISC process was proposed by Ma et al.in 2014, to explain an efficient near-infrared OLED using a donor-acceptor molecule (PTZ-BXP)[26].The molecular structure of PTZ-BXP and the model for exciton relaxation in the electroluminescence process are shown in Figure 2.The estimated efficiency of radiative exciton production was 48%,which is far above the theoretical value of 25%, implying that a large portion of triplet population was harvested to generate electroluminescence.No delayed fluorescence was observed from the transient photoluminescence, indicating that the triplet population were not converted through the TADF or the TTA processes.Quantum chemical computation was performed to explore the possible pathways.The energy gap between the S1and the T1state was calculated to be as large as 0.8 eV so that the reverse ISC process from the T1state to the S1state should be inefficient at room temperature.Nonetheless,the energy of the higher triplet T3state was comparable to that of the S1state, so that the hot ISC process is possibly very fast.Meanwhile, the internal conversion process is relatively inefficient with a large energy of 0.76 eV gap between the T3state and the nearly degenerate T1and T2states.
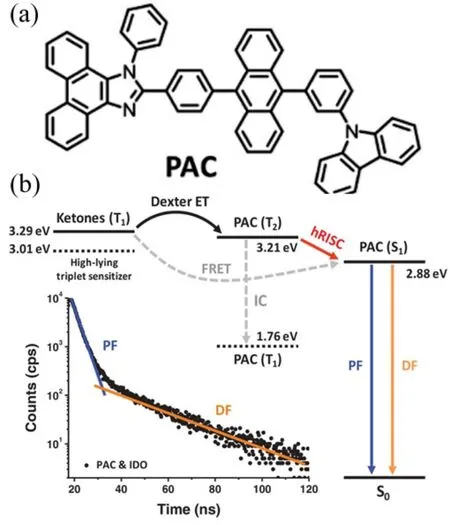
FIG.3.(a) The molecular structure of PAC.(b) Diagram of the energy level alignment of the two sensitizers (ITO and BP) and PAC.The dash arrows stand for the dominant energy transfer process in the ITO-sensitized and the BP-sensitized PAC solution.The dash arrows stand for possible but secondary energy transfer process which were demonstrated negligible in the system.The inset shows the transient decay curve of the photoluminescence from the ITO-sensitized PAC solution.“PF” stands for prompt fluorescence and “DF” stands for the delayed fluorescence attributed to the RISC from the high-lying triplet state (hRISC).Reproduced with permission from ref.29.Copyright 2019 John Wiley & Sons.
To verify the involvement of the hot ISC process,Maet al.[29]carried out the sensitization and probed the carrier dynamics using transient absorption (TA)spectroscopy measurements on an OLED material of PAC (Figure 3).Different triplet sensitizers were employed to populate different triplet excited states.The energy levels of the T1state (1.76 eV) and the S1state (2.88 eV) were deduced from the phosphorescence and fluorescence spectra.The energy level of the T2state was characterized to be 3.21 eV from the excitedstate absorption (ESA) of the T1state.The ESA is induced by the transition from the excited state to the higher excited states.The material of PtOEP was a triplet sensitizer with triplet energy at 1.91 eV, which is used to sensitize the T1state of PAC through the process of triplet energy transfer.Two other sensitizers with different triplet energies, namely IDO (T1:3.29 eV) and BP (T1: 3.01 eV), were used to sensitize the higher lying triplet T2state (3.21 eV) of PAC.At room temperature, both the IDO-sensitized and the BP-sensitized PAC solutions showed delayed fluorescence,whereas at low temperature only the IDO-sensitized sample showed delayed fluorescence.Since the delayed fluorescence is a signature of the reversal ISC process from the sensitized T2state to the S1state,the difference between the delayed fluorescence suggests that the ISC from T2to S1states is the primary channel for fluorescence emission.The direct Frster energy transfer from the T1state of the sensitizers to the S1state of PAC was ruled out.These results confirm the presence of hot ISC process in organic luminescent materials.
II.REVERSAL ISC AND TADF
TADF is from the radiative recombination of the singlet excited states converted from the triplets through the thermally-activated reverse ISC process[18−22].The process converts the “dark” triplet excited states to the emissive singlet excited states, making TADF materials promising for highly efficient LEDs.TADF has gained a rapidly growing interest as a mechanism to improve the efficiency of OLED by harvesting the triplet excitons generated electrically.The first report of TADF in a pure organic molecule was from eosin in 1961[34].In 2011, Adachi et al.reported a pure organic molecule that shows highly efficient TADF[35].OLEDs fabricated using this molecule as emitting layer demonstrated a high EQE value (5.3%), approaching the theoretical limitation of conventional fluorescence molecules.
Typically, triplet excited state has a lower energy than its singlet counterpart.The energy gap between S1and T1(∆EST) is around 0.5∼1 eV[21], making the RISC process energetically unfavorable.Therefore,minimize ∆ESTis of critical importance to harvest back the “dark” triplet excited states to the emissive singlet state.Many organic materials exhibiting efficient TADF have been designed in the framework[36−39].In 2012,Adachiet al.realized highly efficient OLEDs based on a series of metal-free TADF molecules with emission colors raging from sky-blue (473 nm) to orange (577 nm)[21].The molecules were based on carbazolyl dicyanobenzene (CDCB), with carbazole as a donor and dicyanobenzene as an electron acceptor (Figure 2a).The peripheral carbazolyl groups were markedly distorted from the dicyanobenzene plane due to steric hindrance.As a result, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are spatially separated, localized on the donor and acceptor units, respectively (Figure 2b).The spatial separation of HOMO and LUMO orbitals leads to a small overlap between the wavefunctions of the electron and hole,resulting in a small exchange integral and a small singlet-triplet energy difference ∆EST.The ∆ESTof the 4CzIPN, one of the series of CDCB-based molecules, was estimated to be 83 meV by the temperature-dependent transient fluorescence experiments.Using the CDCB-based molecules in multilayer OLED devices, a very high external quantum efficiency of fluorescence emission (19.3%) was achieved for the green organic LEDs (4CzIPN), whilst the orange and sky-blue organic LEDs showed EQEs of 11.2%and 8.0%, respectively.
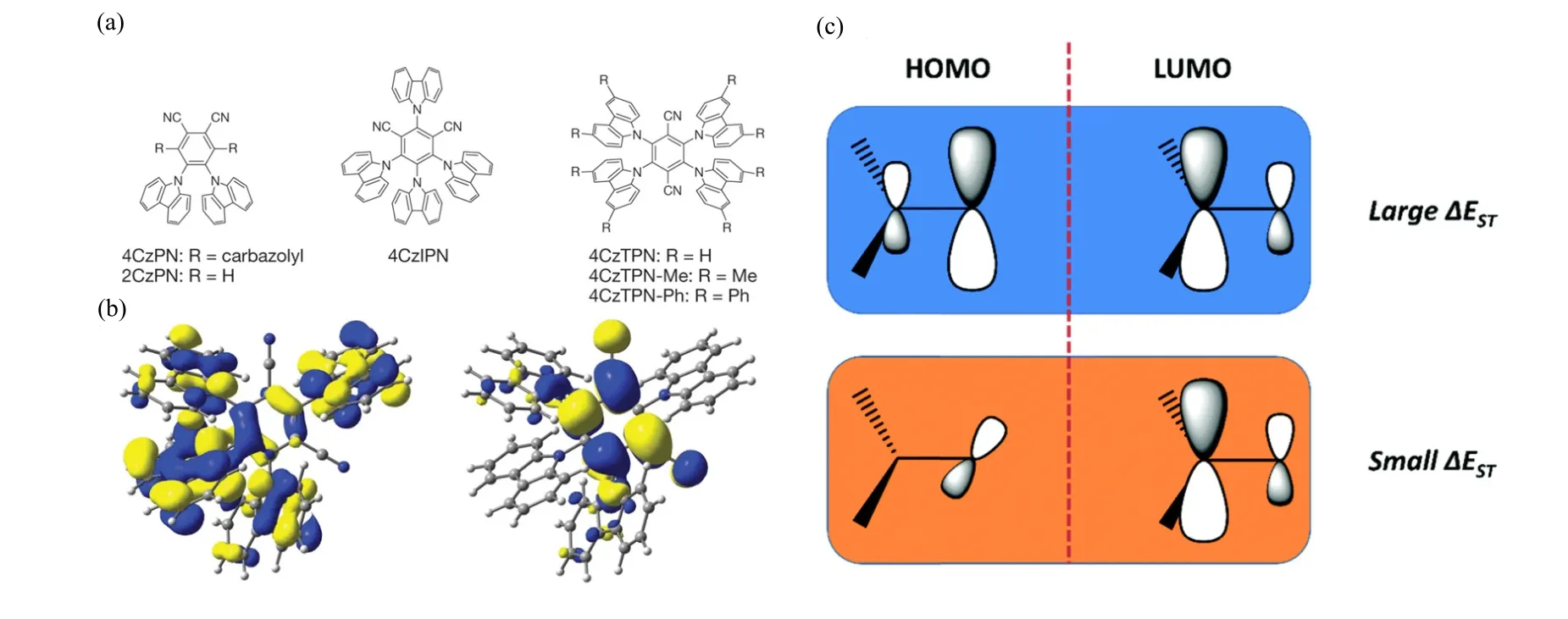
FIG.4.(a)The structure of the CDCB-based TADF molecules.(b)The HOMO and LUMO orbitals of 4CzIPN.Reproduced with permission from ref.21.Copyright 2012.Nature Publishing Group.(c) The molecular design strategy for realizing small ∆EST.Reproduced with permission from ref.22.Copyright 2017.Royal Society of Chemistry.
In the CDCB-based TADF molecules, there is a large dihedral angle between the planes of donor and acceptor.The spatial separation of HOMO and LUMO orbitals (Figure 2b) suggests that the S1state has a charge-transfer (CT) character (1CT)[21].The lowest-lying triplet T1state may be configurated as locally excited triplet (3LE) state or CT triplet (3CT)state.The energy of3CT state is strongly dependent on the electron-withdrawing ability of the acceptor unit and the electron-donating ability of the donor unit[21].The energy of the3LE is insusceptible to these effects[40].The energy landscapes of3CT and3LE states are thus controllable by the electron-withdrawing/electrondonating ability of the donor/acceptor units.If the energy differences between different excited species (the3LE,3CT and1CT)are smaller or comparable to thermal activation energy, the3LE state lying below the1CT state may participate in the TADF process.It is experimentally confirmed that the3LE-3CT splitting is a critical parameter for the rate of TADF in molecules of similar other control parameters[40].These results imply that the3LE state is the intermediate state for the transition between the1CT and3CT states(Figure 5).
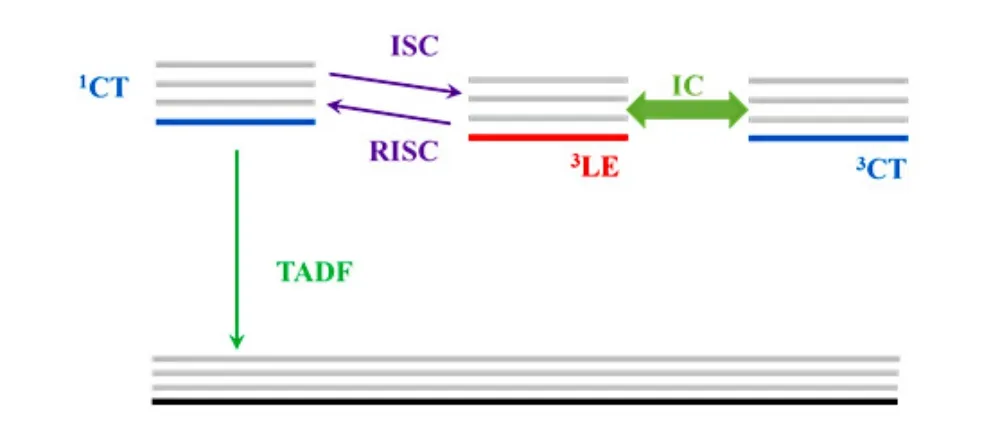
FIG.5.Illustration for the spin-vibronic coupling mechanism for TADF.The interconversion between the 1CT state and the 3CT state is mediated by the 3LE state.
The above discussion is mainly based on the ISC process induced by spin-orbital coupling.According to the EI-Sayed rule[23], the spin-flip transition between the states with different orbital types is much more efficient.The ISC process between the1CT state and the3CT state is much slower than that between the1CT state and the3LE state[41−42].The intermediate of3LE state perfectly bridges the1CT state and the3CT state (Figure 3).This mechanism was demonstrated in many TADF systems both theoretically and experimentally[24,42−44].In the ideal case, the3LE state is on-resonance with the CT state,giving high efficiencies of reversal ISC and hence TADF.It is worth noting that the excited states in the film of organic molecules may be configurated with excimer form with mixed LE and CT characters.The mixed molecule orbital natures possess non-zero integral, resulting in efficient transition between excimer singlet and triplet states without violating the El-Sayed rule[29].In addition to the scenario of spin-orbit coupling, the reversal ISC is also possibly induced by other mechanisms including the hyperfine coupling for the case with small ∆EST[24,42]and the spin-vibronic coupling mechanism[24,42−44].
III.STABILIZED TRIPLETS AND ORGANIC LPL
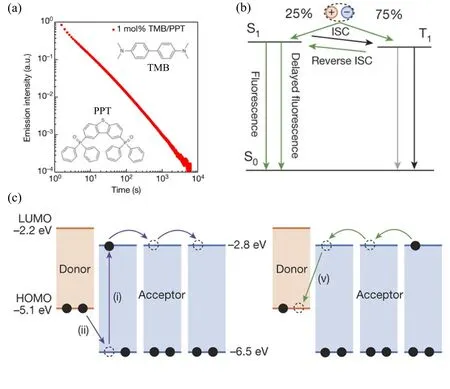
FIG.6.(a) Logarithmic plot of the emission decay profile of a 1 mol% TMB/PPT film.The inset shows the molecular structure of the donor (TMB) and the acceptor (PPT).(b) Triplet harvesting in exciplex emission.Charge recombination produces CT states in a ratio of 25% 1CT states to 75% 3CT states.A small energy gap between 1CT state and 3CT state enables RISC via thermal activation.(c) The proposed emission mechanism of OLPL.Left, the generation of CS states.Right, the recombination of CS states via the diffusion of the electrons among PPT molecules.Reproduced with permission from ref.31.Copyright 2017 Nature Publishing Group.
Organic LPL is the phenomenon when, after being excited, an organic material emits light for several seconds, minutes, or even hours[20,45].Organic LPL has a much longer lifetime compared to the fluorescence, TADF and phosphorescence.Organic LPL can be resulted from the radiative recombination of longlived triplet excitons or the electrons and holes stored in traps[20].For organic LPL based on the long-lived triplet states, stabilizing the triplet excited species and minimizing the non-radiative recombination are essential.In the past few years, several organic LPL systems have been realized, including multi-component guest-host material systems[30], single-component small molecules[46−48], polymer[46], carbon dots[50]and metal-organic frameworks (MOF)[51−54].
To further extend the persistence of organic luminescence, Adachi and Kabe introduced an organic donor-acceptor blend system in which the diffusiondriven charge recombination process induced the persistent luminescence[31,55−58].Strikingly, the organic LPL lasted for more than 1 hour in the guest/host system (Figure 6).The emission spectrum of the organic LPL is similar to that of the steady fluorescence,which is broadened and featureless, indicating that the organic LPL was from the radiative combination of the charge-transfer states.The decay profile of the LPL followed the Debye-Edwards law because of the presence of the intermediate states[31,56,59].It is suggested that the CT states formed via photoexcitation undergo the charge separation process to generate free carriers.Because the donors were dispersed discretely among the acceptors, the diffusion of the electrons was more efficient than that of the holes.When the electrons and holes encountered at the interface of donor and acceptor after the slow diffusion process, the interfacial1CT states and3CT states would be formed in 1:3 ratio.Organic LPL was generated through the radiative recombination of these CT states.The3CT states may be harvested through the reversal ISC process.
When the energy of the3LE state localized on the donor or the acceptor molecule is lower than that of the CT state,energy transfer from the CT state to the3LE state may take place.The energy transfer process between the CT state and the3LE state was observed in a blend system with a small molecule (TMB) as donor and a polymer (PBPO) as acceptor[58].The energy of the3LE state of the donor molecule is lower than that of the CT state based the comparison between the CT emission from the blend and the phosphorescence from TMB.In addition to the organic LPL emission with the decay profile following Debye-Edwards law,an additional exponential-decay component in the first 10 seconds was observed due to the phosphorescence from the3LE state.The spectrum of the organic LPL emission resembles that of the TMB phosphorescence,indicating that the organic LPL was mainly originated from the3LE states of the donor rather than the CT states.Adachi et al proposed a mechanism for the organic LPL in this doped polymer system as shown in Figure 7.Firstly, the1CT states and donor3LE states were generated from the donor1LE states through the energy transfer and ISC process, respectively.An equilibrium between the nearly degenerate1CT states and the3CT states was then established through the ISC and reversal ISC process.Most3CT states was converted to the3LE states through the energy transfer process with the3LE states below the3CT states.Small portion of the CT states underwent the charge separation process to form the CS states.Slow recombination of CS states generated1CT states and3CT states in 1:3 ratio, followed by energy transfer process from the3CT states to the3LE states.Both3LE states and the CT states contributed to the organic LPL with phosphorescence from the3LE states in the majority.
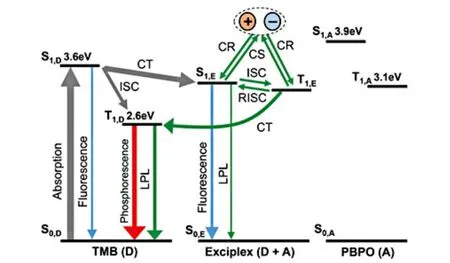
FIG.7.Jablonski diagram for a TMB/PBPO film.Charge recombination generates 1CT and 3CT states in a 1:3 ratio.The 3CT states can be converted to the donor 3LE states through the IC process.OLPL is contributed of the emission from the 1CT states and the phosphorescence from the donor 3LE states.Reproduced with permission from ref.58.Copyright 2018 John Wiley & Sons.
The energy gap between the1CT states and the3LE states can influence the performance of organic LPL emission.Adachiet alcompared three donoracceptor blend systems with different energy gaps between the1CT states and the3LE states[56].It was found that materials with small energy gaps showed longer emission time duration.As for the material with large energy gap between the1CT state and the3LE state, emission from the1CT states and the3LE states contribute to the organic LPL.
In the donor/acceptor blend systems, the CS states have been suggested to account for the exceptionally long-persistent emission.However, the formation mechanism of such a CS state in organic LPL materials remains elusive.The clarification of the mechanism behind would be helpful for the rational design of new organic LPL materials.To better understand the mechanism of organic LPL,our group used the time-resolved optical spectroscopy together with the electron spin resonance (ESR) spectroscopy and quantum chemistry computation to reveal the excited states dynamics upon photoexcitation in a model system TMB/PPT[61].It was found that the intermediate triplet states with mixed local excitation and CT characters bridge the photo-excited1CT states and the CS states, enabling the LPL in the organic LPL system (Figure 8 (a)).
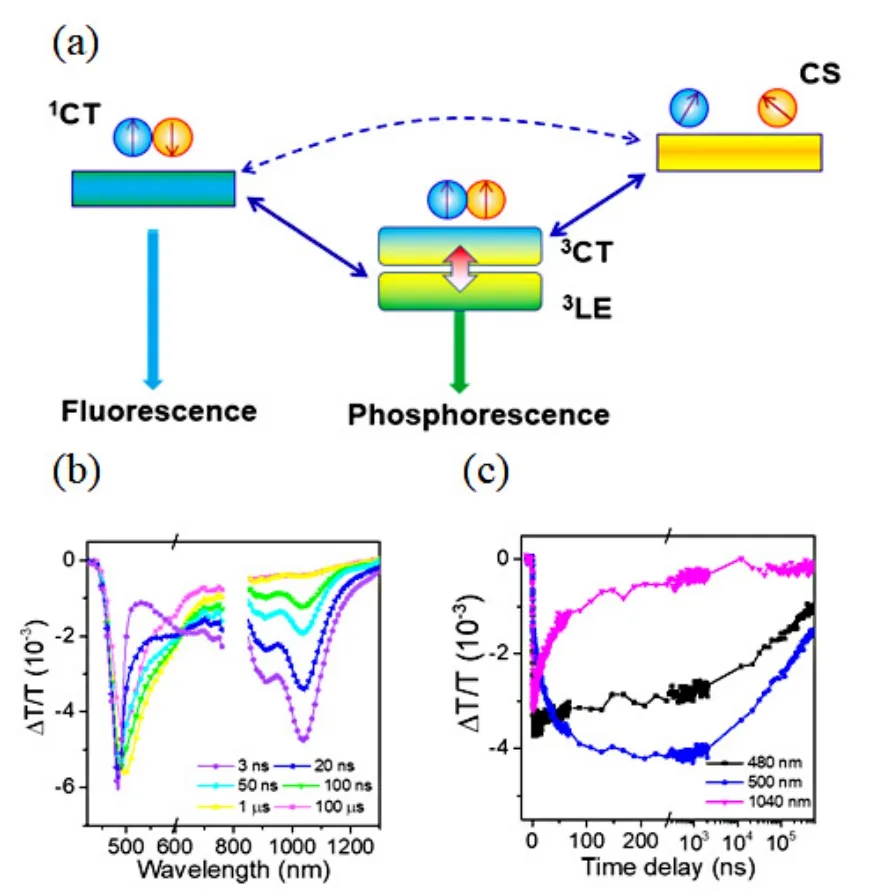
FIG.8.(a) Diagram of charge separation through intermediate triplets of mixed 3LE and 3CT states.(b)Ns-resolved TA spectra of a 10 mol% TMB/PTT film.(c) Ns-resolved kinetic curves probed at the different wavelengths.Reproduced with permission from ref.61.Copyright 2020 American Chemical Society.
The ISC process with a lifetime of 52 ns from the photo-generated1CT state to an intermediate triplet state was observed by ns-resolved TA measurements(Figure 8 (b)&(c)).The generation of the intermediate triplet states was also confirmed by the ESR and the time-resolved fluorescence measurements.To figure out the nature of the intermediate triplet states,we explored the possible triplet configurations and their energy landscapes by quantum chemistry computation.It was found that the3LE state at TMB is the lowest triplet excited state and the energy gap between3LE state at TMB and3CT is around 30 meV.According to the EI-Sayed rule, the ISC process between the1CT and3LE state is much more efficient than that between the1CT and3CT state.Therefore, the process with a characteristic lifetime of 52 ns was assigned to the ISC process from the1CT state to the3LE state at TMB.The small energy difference between the3LE state at TMB and the3CT state allows efficient mixing between them at room temperature.These findings suggest that the triplet intermediate is probably a mixture of the3LE and3CT states, which is consistent with the result that the ESA feature was similar to, but not identical as that of the3LE state at TMB.We monitored the charge separation from the intermediate triplet state by TA measurements with temporal window covering from milliseconds to seconds and observed the unbalanced diffusion of TMB cation and PPT anion.The involvement of the intermediate triplet state in the organic LPL was confirmed by the temperature-dependent LPL measurement.When the temperature decreases, the spectra of the LPL emission get much more structured and an additional exponential-decay component was observed, indicating a higher3LE population at low temperatures.The ratio between the LPL and the phosphorescence decays significantly,which supports the scenario of charge separation from3CT states in the blend.
IV.CONCLUSION AND OUTLOOK
Triplet excited states play a vital role in promoting the luminescent emission in organic materials.Conventionally, the triplet state is considered as dark for fluorescence.Nevertheless, the triplet state serves as an intermediate that converts the triplet population to fluorescent singlets.The role of triplet state is highly system dependent and dramatically different at different stage of carrier dynamics.The hot ISC process is desirable when the coupling between hot excited state is sufficiently strong to compete with the internal conversion process.The reversal ISC is highly efficient when the3LE state bridges the low-lying singlet and triplet excited species.The stabilized triplet state induces LPL emission at a late stage.These processes can be implanted to demonstrated cost effective optoelectronic applications including the LEDs,sensors and imaging agents.In comparison with the inorganic counterparts, organic materials have the advantages such as high structural diversity and property tunability, large active areas, mechanical flexibility and low-cost processing[20].So far, most studies on the triplet dynamics are based on time-resolved emission spectroscopy and quantum chemical computation.Together with the material design, the dynamics of triplet excited species are established from indirect experimental evidences.Ultrafast transient absorption spectroscopy and time-resolved electron spin resonance measurements may provide more insights into the exact dynamics and kinetics of specific species of different excited states.Moreover, other spin-related processes including singlet fission [60]and doublet excited states [62]have also been proposed to develop organic emitters.Further study on the excited states of different spin configurations is instrumental for understanding the physics and developing state-of-art light emitters.
ACKNOWLEDGMENTS
This work is supported by the National Key R&D Program of China (Grant Nos.2018YFA0209101 and 2017YFA0303700), the National Science Foundation of China(Grant NOs.21922302,21873047,91833305,and 91850105)
