超极化核磁共振方法的原理和应用
2020-04-24庞振峰管晗曦高李娜曹伟成尹竟琳孔学谦
庞振峰,管晗曦,高李娜,曹伟成,尹竟琳,孔学谦
浙江大学高新材料化学研究中心,化学系,杭州 310027
1 引言
核磁共振(nuclear magnetic resonance,NMR)是一种尖端的化学分析和医学检测技术。在化学、材料学、分子生物学领域被广泛应用。同时,凭借其非侵入性检测的特点,磁共振成像技术在基础医学和临床诊断中扮演了关键角色。虽然核磁共振功能强大,但是较低的检测灵敏度一直是技术发展的瓶颈。核磁共振信号来源于原子核与外加磁场的塞曼作用,其信号强度正比于原子核在不同能态的布居数之差,也称为极化,polarization,P。例如自旋1/2原子核处于热平衡时,能态的分布满足玻尔兹曼分布,此时极化为
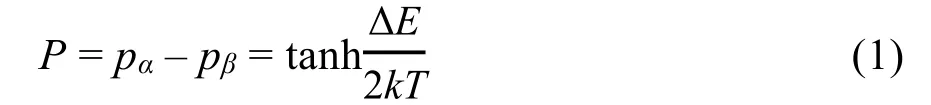
可以看出信号强度取决于两个能态的能量差,ΔE,和温度,T。能量差,ΔE = Eβ− Eα= γħB0由自旋的旋磁比和外加磁场的强度决定。以常见的9.4 T磁场为例,室温下极化P ≈ 3 × 10−5。与紫外可见吸收光谱(P ≈ 1)和红外光谱(P ≈ 0.6)相比核磁共振的灵敏度是极低的。
为了增强核磁共振的信号,人们可以施加更高的磁场(增大ΔE),选择较低的温度(降低T)或使用低温探头(减小热噪声,增加信噪比)进行实验。但一般来说常用的核磁共振设备能够检测到最低约1015个核自旋信号,这相对于其他光谱技术来说相差了很多个数量级1。为了进一步提高核磁共振的灵敏度,人们试图通过一系列化学或者物理方法打破原子核塞曼作用的热平衡状态,从而原子核极化有数量级的提升,这类方法统称为超极化(hyperpolarization)技术。1953年,Slichter等人利用电子极化转移的方法,成功实现了7Li原子核的超极化2,此后的数十年中多种超极化技术应运而生。现阶段的超极化技术可分为两类(图1)3,一类是通过将电子自旋的极化转移到原子核来实现原子核的超极化(其中电子极化又可分为热平衡态极化和电子的超极化);另一类是利用特殊的分子极化来实现原子核的极化。
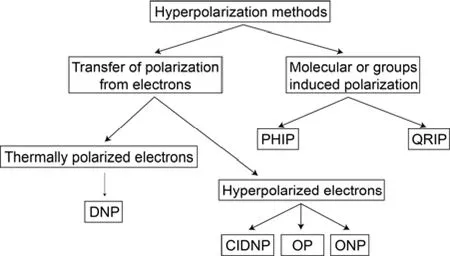
图1 根据极化来源对超极化方法进行分类3Fig. 1 Hyperpolarization methods grouped by polarization sources 3.
电子具有较大的旋磁比(gyromagnetic ratio),热平衡态的电子极化是相同温度下热平衡质子极化的660倍。所以,即使将处于热平衡态下的电子极化转移到原子核上也会很大的提高原子核的极化。动态核极化技术(Dynamic Nuclear Polarization,DNP)就是利用热平衡电子极化来增强原子核极化的技术。另外,我们可以通过激光技术或化学反应实现电子的超极化,然后将超极化电子的极化转移,以实现更高的原子核极化。光泵(Optical Pumping,OP),光核极化(Optical Nuclear Polarization,ONP)和化学诱导动态核极化(Chemically induced dynamics nuclear polarization,CIDNP)都是利用超极化电子实现原子核超极化的技术。还有一些比较特殊的分子(比如氢分子)或者化学基团由于原子核之间的强耦合,在低温低磁场下会形成单一自旋态。这些特殊自旋态转变之后也可以打破核自旋的热平衡分布,实现原子核的超极化,例如仲氢诱导极化(Para-hydrogen induced polarization,PHIP)和量子转子诱导极化(Quantum rotor induced polarization,QRIP)4。这篇综述主要介绍几种常见超极化技术的原理及应用。
2 动态核极化
动态核极化是一种可显著提高核磁信号强度的超极化方法。1953年, Overhauser首先提出通过激发金属中的自由电子,实现电子到原子核的极化转移5。之后Carver和Slichter在低磁场(3 mT)下用7Li证实了该理论2。DNP是用连续微波照射样品,使电子跃迁饱和,通过电子和原子核的耦合作用,改变核自旋能态的分布使其极化大大增强,从而提高核磁共振信号强度。以1H核为例,理论上信号可增强γe/γH≈ 660倍。如此显著的灵敏度增强效应使DNP方法在液体和固体核磁中均得到了广泛应用。
2.1 DNP极化转移机理
DNP极化转移机理可分为Overhauser效应(Overhauser Effect,OE),固态效应(Solid Effect,SE),热混合效应(Thermal Mixing,TM)和交叉效应(Cross Effect,CE)四种机理。
2.1.1 Overhauser效应
Overhauser效应主要适用于低磁场下的液体、含自由电子的金属及有机导体样品。这些样品中电子运动自由度大,转动相关时间(τ)短。在低磁场下可以满足条件ωeτ < 1,其中ωe是电子的Larmor频率。此时电子的极化可以通过交叉弛豫(Crossing Relaxation)有效的转移到与其存在超精细相互作用的原子核上。图2a为一个自由电子和一个自旋1/2的原子核在磁场下组成的系统。系统由βeαn,βeβn,αeαn,αeβn(电子态在前,简单表示为λ1,λ2,λ3,λ4)四个能级组成。实验过程中,先用微波饱和电子(pλ1= pλ3, pλ2= pλ4, pλi为每个能级的布居数)。在各向同性超精细耦合产生的交叉弛豫作用下pλ1增大pλ4减小,最终导致原子核能级间布居数差异(pλ1+ pλ3) − (pλ2+ pλ4)增大,核极化增强。
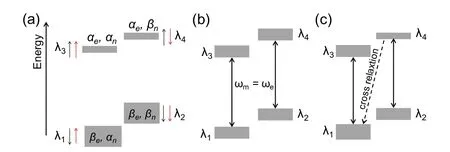
图2 Overhauser效应的能级分布图。(a)热平衡态下四个能级的能量及布居数(黑色和红色箭头分别表示电子和核的自旋方向,灰色方块大小代表该能级的粒子数多少)。(b)微波照射下电子的饱和跃迁对应的各能级的布居数分布(微波频率等于电子Larmor频率,ωm = ωe)。(c)交叉弛豫作用下(λ4 → λ1)各能级布居数分布Fig. 2 Overhauser effect: (a) Schematic representation of four energy levels and their populations (size of gray bars)at thermal equilibrium. (black and red arrows indicate electron spins and nuclear spins, respectively)(b) Population distribution with the electron saturation transition at the microwave irradiation (ωm = ωe).(c) Population distribution with the cross relaxation.
理想的OE必须满ωe<< 1,保证交叉弛豫λ4→ λ1的速率远大于电子弛豫(λ3→ λ1,λ4→ λ2)的速率,从而获得高的极化转移效率。一般来说只有在低磁场的液体或导体样品中才能通过OE高效转移极化。固体材料的DNP增强主要通过固态效应(SE)、热混合效应(TM)和交叉效应(CE)来实现。固态效应作用机理主要来源于电子和核的超精细耦合作用,交叉效应既有电子之间的耦合也包含了电子和核的超精细耦合,热混合效应则可以看作交叉效应和电子间自旋扩散的组合。
2.1.2 固态效应
固态效应发生在低温下含有固定顺磁中心的固体样品中,此时未成对电子定域在某个原子或晶格中不可移动。固态效应要求电子EPR谱线宽度小于原子核的Larmor频率,此时电子–原子核自旋体系在较强的耦合作用下成为一个包含混合态的四能级系统。新的混合态由原来的λ1和λ2,λ3和λ4能级混合得到(图3)。混合程度系数q受到电子与原子核之间的耦合强度和外磁场强度影响。混合态的存在可以使禁阻的零量子和双量子跃迁在有效微波照射下被激发。当微波照射频率为ωe+ ωn时,可激发λ2*能级到λ3*能级的双量子跃迁,由于电子的弛豫速率远大于核,因此受激跃迁后λ3*态快速弛豫λ1*态,通过不断的激发-弛豫过程最终导致与电子相邻的原子核在α与β能级的布居数差异增大,获得正向的DNP增强效应。同样地,当微波照射频率为ωe− ωn时,可激发λ1*能级到λ4*能级的零量子跃迁,获得负向的DNP增强效应。
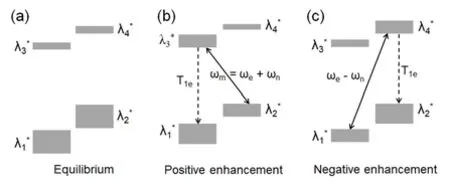
图3 固态效应的能级分布图:(a)热平衡态下四个混合态能级的能量及布居数(λ1* = λ1 + qλ2,λ2* = λ2 − qλ1,λ3* = λ3 − qλ4,λ4* = λ4 + qλ3,q 为混合程度系数)。(b) ωe + ωn微波照射下的能级跃迁及电子弛豫引起的正向增强。(c) ωe − ωn微波照射下引起的负向增强Fig. 3 Solid effect: (a) Schematic representation of four energy levels and their populations (size of gray bars) at thermal equilibrium. (λ1* = λ1 + qλ2,λ2* = λ2 - qλ1,λ3* = λ3 − qλ4,λ4* = λ4 + qλ3, q indicates mixing factor)(b) Microwave irradiation at ωe + ωn leads to positive enhancement. (c) Microwave irradiation at ωe − ωn leads to negative enhancement.
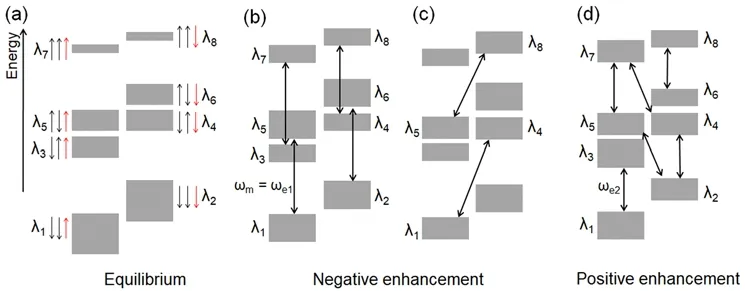
图4 交叉效应的能级分布图。(a)热平衡态下八个能级的能量及布居数(黑色和红色箭头分别表示电子和核的自旋方向)。(b) ωe1微波照射下电子1的饱和跃迁及(c)电子2和核的零量子跃迁对应的各能级布居数分布,最终产生负向增强效应。(d) ωe2微波照射下电子2的饱和跃迁及电子1和核的双量子跃迁最终产生正向增强效应。Fig. 4 Cross effect: (a) Schematic representation of eight energy levels and their populations (size of gray bars)at thermal equilibrium. (black and red arrows indicate electron spins and nuclear spins, respectively)(b) Saturation of the allowed EPR transitions for electron 1. (c) Zero quantum transition of the electron 2 and nucleus leads to negative enhancement. (d) Saturation of the electron 2 and double quantum transition of the electron 1 and nucleus leads to positive enhancement.
2.1.3 交叉效应(CE)
当电子顺磁共振(EPR)的线宽大于核的Larmor频率,且EPR的增宽主要来源于g因子的各项异性导致的非均匀增宽时,即Δ > ωn> δ (ωn为核的Larmor频率,Δ和δ分别为电子顺磁共振的非均匀和均匀增宽),DNP增强机理主要为交叉效应。交叉效应利用双自由基为极化试剂,是目前高场实验中最有效的一种DNP增强机理。
交叉效应中包含了电子之间的耦合作用,通常以两个电子和一个核的三自旋体系为模型来研究其作用机制。在外磁场作用下,该三自旋体系裂分为8个能级,其能级图如图4所示。在微波照射下,交叉效应中包含两种能级跃迁:(1)当微波照射频率与其中一个电子自旋共振频率相等即ωm–ωe1或ωm– ωe2时,其中一个电子受激跃迁,两个电子的极化度出现差异;(2)当两个电子的自旋共振频率差与核自旋频率相等即ωe1− ωe2~ ±ωn时,其中一个电子与核的自旋方向同时发生翻转,发生零量子或双量子跃迁(分别导致负向和正向增强)。通过上述两种跃迁可使核极化增大。从而获得增强的NMR信号。
2.1.4 热混合效应(TM)
当体系中自由基浓度较高,电子之间的强耦合作用导致均匀增宽的电子顺磁共振谱线宽大于核的Larmor频率,即δ > ωn时,热混合效应(TM)占主导地位。TM与CE类似,但TM中电子之间的耦合作用更强,我们可以将电子-核自旋系统描述为三个相互作用的热库系统来理解热混合效应。这三个体系分别为电子塞曼系统(EZS),电子偶极系统(EDS)和核塞曼系统(NZS)。其中,每个系统的自旋状态(极化率)可用自旋温度(体系混乱度)来表示。首先,通过一个非共振的微波照射激发部分电子跃迁,在电子顺磁共振谱上产生一个大的极化梯度,这个极化梯度的产生导致EDS系统自旋温度降低。之后,通过EDS和NZS系统之间的热接触(电子与核的耦合作用)降低了NZS系统温度,最后导致核极化的增强。极化增强的具体作用机制取决于实验条件,包括样品性质,自由基种类及浓度等(表1)6。
2.2 自旋扩散
在DNP实验中,对于液体和金属样品,由于分子的快速运动或自由电子的存在,样品中所有的核自旋都能有效地与电子自旋直接作用实现电子到核的极化转移。然而在固体电介质材料中,只有少数与电子相邻的核(中心核)可以与电子有直接耦合作用,从而感受到电子的极化转移,而核磁信号主要来源于与电子相距较远的体相核,因此样品的极化增强要通过以下两个过程来实现:(1)电子极化转移至与其相邻的中心核,(2)通过自旋扩散过程将中心核的极化转移至体相核。自旋扩散效率决定了总的极化增强效率。自旋扩散的效率取决于原子核之间的耦合强度,其受原子核种类和原子核之间的距离影响6,7。
2.3 实验装置及应用
DNP实验的第一次成功应用是在小于1 T的低场核磁中8,且Overhauser效应9,10和固态效应的极化增强效率会随着磁场强度的增强而减弱。随着高频微波源和其他硬件设备的发展,DNP增强实验在强磁场(> 5 T)和魔角旋转条件下都得到了广泛应用11–13。DNP谱仪的基本结构主要包括三部分:微波源(发射微波),波导(将微波传入探头),低温探头(检测核磁信号)。由于低温下自旋弛豫变慢可提高电子到核的极化转移效率,且根据玻尔兹曼分布,温度越低,电子与核在热平衡态的极化度越高,因此大多数DNP实验都在超低温下进行。我们知道,普通核磁实验所使用的探头一般是一个或几个由线圈和电容器组成的谐振回路,而由于电子顺磁共振激发频段较高,一般采用谐振腔进行微波功率的接收。在DNP中,将谐振腔与射频谐振电路中的线圈一体化设计构成双共振探头可用于微波照射和射频信号的发射和接收。同时可从谐振腔底部通入冷的氮气或氦气来进行低温实验。DNP实验中,需要引入极化试剂作为极化源来诱导核极化。对于极化试剂的选择,主要基于以下因素:自由电子顺磁共振频率谱宽,自由基溶解性和毒性,自由基反应活性和弛豫时间等。目前主要以氮氧双自由基为极化试剂14。

表1 四种DNP增强机理对比6Table 1 Four common DNP mechanisms 6.
随着DNP的发展,其在物质结构解析15,生物大分子检测及磁共振成像(Magnetic Resonance Imaging,MRI)16等方面的应用越来越广泛。材料的界面结构和性质会极大地影响其功能和应用,固体核磁技术可以实现原子水平上的表面结构表征,在先进材料的发展方面有着重要的指导意义。然而很多材料具有低的表面积或低浓度的表面活性位点,核磁的低灵敏度往往限制其应用,通过DNP表面增强技术可解决这一问题。将固体样品浸润于有机自由基溶液中,将溶液中的自由基电子极化转移到观测核,可得到普通NMR条件下难以检测的稀核如13C17,15N18等的高分辨谱图,以及在较短时间内获取高分辨二维谱图,从而实现对其表面结构的解析。DNP表面增强技术在功能化材料19,金属有机框架材料(MOFs)20,纳米颗粒以及多孔材料21的表征方面都有广泛应用。在生物大分子的检测方面,通过极化增强的15N及二维谱可解析膜蛋白和多肽的结构信息以及它们与脂质分子的相互作用22,23。基于DNP的成像技术可实现在活体内对重要的生物小分子成像,捕捉代谢物信息,同时DNP可实现稀核(13C)在MRI中的检测,在短时间内获得高分辨图像24。
3 光泵
光泵(Optical Pumping, OP)是一类利用光来实现原子核超极化的方法。现阶段使用的光泵技术可分为两类,一类是间接光泵法,另一类是直接光泵法。间接光泵法通过自旋交换光抽运(Spinexchange optical pumping,SEOP)进行介绍,直接光泵法通过亚稳态交换光抽运(Metastableexchange optical pumping,MEOP)进行介绍
3.1 自旋交换光抽运原理
自旋交换光抽运是一种重要的间接光泵方法。它以碱金属气体为媒介利用圆偏振光实现稀有气体原子核的超极化(图5a)25。考虑到碱金属的蒸汽压和自旋交换速率等因素,铷是最适合SEOP的碱金属26。铷的基态电子构型为[Kr]5s1,对应基态52S1/2,吸收一个光子后子5s电子激发到5p轨道上,由于电子的自旋-轨道耦合作用(J = L + S)占据5p轨道的激发态分裂为两种能态(精细结构),第一激发态52P1/2(J = 1/2),和第二激发态,52P3/2(J = 3/2)。第一激发态和第二激发态的激发能量分别为D1=795 nm,D2= 780 nm。铷主要有两种天然同位素85Rb (72.2%,I = 5/2)和87Rb (27.8%,I = 3/2),原子核与电子的自旋-自旋耦合作用(F = I + J)会产生超精细结构(图5b)27。对于基态和第一激发态都存在两种超精细结构,F = I + 1/2和F = I − 1/2。当存在外加磁场的时候,超精细结构的能态由于塞曼效应会分裂为2F + 1个能态。例如对于85Rb的第一激发态52P1/2(J = 1/2),超精细结构F = 5/2 ± 1/2,对于F=2的超精细结构在磁场中存在5个能态(mF=−2,−1,0,1,2),对于F = 3的超精细结构在磁场中则存在7个能态(mF= −3,−2,−1,0,1,2,3)。
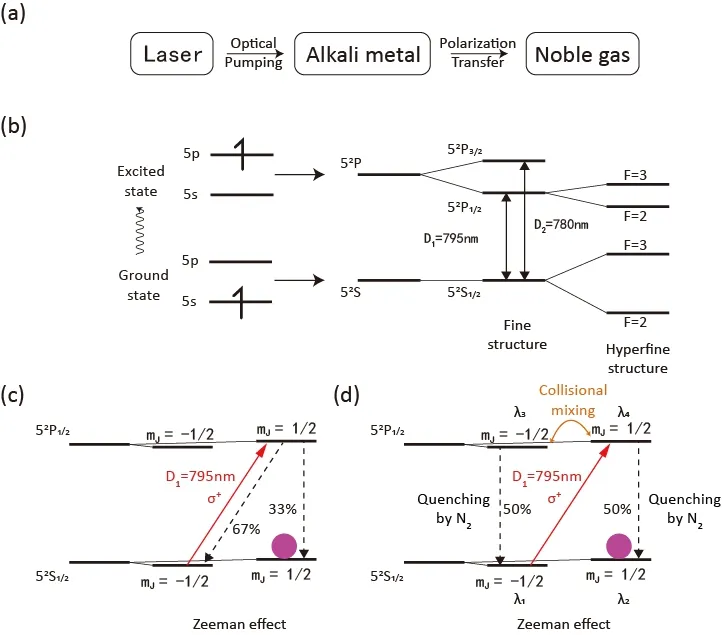
图5 (a) SEOP的极化产生与转移25。(b) 85Rb的光谱结构27。(c)合理忽略超精细结构后85Rb在左旋圆偏振光的照射下发生极化25。(d)实际实验条件下85Rb发生极化25。Fig. 5 (a) Process of spin-exchange optical pumping 25. (b) Orbital structure, fine structure and hyperfine structure of 85Rb 27. (c) 85Rb is hyperpolarized with irradiation of left circularly polarized light (hyperfine structure is ignored because of pressure broadening) 25. (d) 85Rb hyperpolarization in the presence of N2 25.
由于超精细结构和塞曼效应极其复杂的谱线,实验中会选择合适的气体压力利用压力增宽(pressure broadening)效应使超精细谱线不能分辨,并使用低磁场使超精细耦合下的塞曼效应弱于超精细耦合。碱金属基态超精细结构的能级差都小于10 GHz,在常用的实验条件下(> 1 amagat,~0.002 T)碱金属原子的吸收宽度在30 GHz左右,超精细结构和塞曼效应的影响可以被合理忽略28。
简化后的体系只存在精细结构,不需要考虑原子核的影响,在外加磁场下每个精细结构的能态会裂分为2J + 1个能态。当选用波长为795 nm的左旋圆偏振光沿外加磁场方向对基态铷原子进行激发时,只有λ1→λ4满足跃迁选律Δmj= +1, ΔL = ±1,ΔJ = 0,±129。处于λ4激发态的铷原子会有2/3的概率回到λ1,1/3的概率回到λ2,回到λ1的铷原子会继续被激发到λ4然后一部分回到λ1,另一部分回到λ2;回到λ2的原子不会被左旋圆极化光激发,其布居数会不断积累,这样就完成了铷原子的超极化(图5c)25。
在实际情况中,一部分铷原子会以辐射光子的形式从激发态回到基态,而辐射出的光子是不具有偏振性的,当铷原子蒸汽的密度较大,这种光子会有较大几率被铷原子重新吸收。不具有偏振性的光子可以把处于λ2的铷原子也激发,循环往复不能使铷原子在某一个特定的态上积累,从而不能实现超极化,这种现象被称为辐射捕获(radiation trapping)。为了减少辐射捕获的发生,体系中会加入氮气来稀释铷原子蒸汽。在氮气分子和铷原子的碰撞过程中,激发态的铷原子会将能量转移给氮气分子回到基态而不会辐射出光子,转移的能量变为氮气分子的振动能30。实验中除了会加入氮气还会加入4He作为缓冲气,氮气和4He会使λ3和λ4两个能态发生碰撞混合(collisional mixing),碰撞混合会使两个态发生快速交换从而使两个态的布居数相等,这样就会使激发态的铷原子,有1/2的概率回到λ2基态,比没有碰撞混合时的1/3更为高效(图5d)25。
碰撞混合同样也会发生在基态λ1和λ2之间,但是由于s轨道的球对称性会使两个基态能态的混合远小于对称性差的激发态轨道(p轨道)。极化后的铷原子如果与容器壁发生碰撞也会造成两个基态能态的混合降低极化。把能引起两个基态能态混合,使极化降低的所有因素综合考虑,使用自旋破坏速率(spin-destruction rate),ΓSD,来表征极化降低的过程,其数值越大表示极化衰减的越快。处于稳态(足够长的时间照射,各个态的布居数稳定)的铷原子的极化可表示为,其中PRb表示稳态铷原子的极化,γopt为光泵速率(受到光流密度和光子吸收概率影响),ΓSD为自旋破坏速率。从上式中可以得出,要想提高铷原子的极化需要增大光泵速率降低自旋破坏速率。

得到了极化的铷原子,下一步就是要把铷原子的极化转移给稀有气体原子核。129Xe凭借其较高的天然丰度(26.44%)和与铷原子较快的自旋交换速率,在SEOP中被广泛使用。铷原子与129Xe原子核的自旋交换(spin-exchange)是通过碰撞完成的。129Xe的稳态极化可以表示为,

其中PXe为129Xe的稳态极化,PRb为铷原子蒸气的稳态极化,γSE为铷原子与129Xe原子核的自旋交换速率(受气体压强和铷原子浓度影响),Γ为129Xe的弛豫速率(主要由129Xe互相之间的碰撞和129Xe与容器壁的碰撞造成)31–34。
3.2 亚稳态交换光抽运原理
亚稳态交换光抽运是一种直接光泵的方法,利用光直接极化被照射的原子核。这里以3He(I = 1/2)为例介绍低磁场下亚稳态交换光抽运的原理35–37。
氦原子的核外电子基态构型为1s2,基态为11S0,由于此基态的J = 0不会因为核自旋的改变而出现不同的能态,也不会受到塞曼效应的影响。所以无法通过此基态来完成原子核或电子的超极化。当处于基态的氦原子经过射频放电(RF discharge)后电子构型变为1s12s1,由于2s的电子回到1s是跃迁禁阻的,这种态会有较长的寿命所以被称为亚稳态(metastable state),23S1。亚稳态23S1吸收一个光子(1083 nm)后根据跃迁选律ΔL = ±1,ΔJ = 0,±1会得到三种激发态23P0,1,2(图6a)。考虑到3He的超精细耦合和外加磁场的塞曼效应,3He的能级分布如(图6b)38。
C8和C9是低场光泵中最高效的两条谱线38。23S1和23P0在磁场和超精细耦合作用下分别存在6个和2个能态分别记为A1到A6和B17,B18。A1到A6的波函数可写成纯态│mJ, mI>的叠加形式,mJ描述电子的行为,mI描述原子核的行为,其中A1= │−1,−>和A4= │1, +>是两个纯态。在左旋圆偏振光的作用下ΔmF= 1,A4态的布居数会得到积累。若3He原子在A4态上富集,原子核的自旋态也会在│+>态上富集,从而实现核极化(图6c)25。类似的,在右旋圆偏振光的作用下ΔmF= −1,A1态的布居数会得到积累,原子核在|—〉态上富集,也会实现核极化。计算表明使用右旋偏振光具有更高的核极化效率38。

极化后的3He原子仍旧处于亚稳态,亚稳态的原子可以与基态的原子通过碰撞来相互转化。极化的亚稳态He*+转变为极化的基态He+,未极化的基态He转变为未极化的亚稳态He*然后被光泵极化为He*+。循环往复可以将未极化的基态He转变为极化的基态He+。

图6 (a) He原子基态、亚稳态、第一激发态能级图。(b) 3He原子亚稳态和第一激发态超精细结构38。(c)以C8谱线为例,3He亚稳态在左旋圆偏振光的作用下发生超极化25。Fig. 6 Level diagram of (a) He including ground state, metastable state, and excited state;(b) hyperfine structure of metastable state and excited state of 3He 38.(c) hyperpolarization of 3He with the irradiation of C8 line 25.
3.3 半导体光抽运(Optical pumping of semiconductors)
在一些半导体中也可以实现光泵39,40。位于价带或导带边缘的电子占据位点可以分别被看作原子能态一样的2P3/2和2S1/2。在磁场和左旋圆偏振光的作用下电子也会发生极化。极化的电子通过费米接触(Fermi contact)作用和与原子核的偶极作用可以进行极化转移,实现核的超极化(图7)25。
3.4 实验装置与实验条件
SEOP的实验装置如图827所示27,129Xe,4He,N2混合气通入加入液体铷的圆柱状石英容器中,容器中的均匀磁场和圆偏振光发生装置都沿轴向。装置配有控温系统可用来调节圆柱状容器中气体的温度。装置内有磁共振线圈可用来检测129Xe的极化情况。实验中需要对圆柱形容器的高和直径进行合理选择,尽量缩短极化后129Xe到储存装置的路径长度,以减少129Xe与容器壁碰撞产生弛豫。激光的功率和波长、圆柱形容器的温度、极化时间和129Xe的分压都需要合理选择以获得最大极化率41。SEOP的磁场强度一般在10 mT左右,气体极化室尺寸为长度~30 cm、直径~5 cm,温度保持在~400 K,混合气的体例为(~2%氙气(天然丰度或129Xe),~10%氮气,~88%氦气),气体的总压力在几个大气压左右。MEOP的实验装置与SEOP的实验装置类似,但是不需要碱金属蒸气,并且整个实验中都需要持续的弱射频放电来保持亚稳态3He的数量。低磁场下的MEOP需要3He的压强在1mbar的量级,以减少碰撞产生的能态混合,从而造成极化损失。因为3He极大的扩散系数,MEOP对磁场的均匀度要求更高(ΔB/B ~ 50 ppm∙cm−1,B < 30 mT)42。
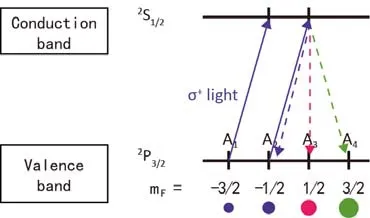
图7 半导体光抽运中的电子极25Fig. 7 Scheme of optical pumping of semiconductors 25.

图8 气体连续流动85Rb-129Xe SEOP装置图27Fig. 8 Scheme of continuous-flow apparatus for 85Rb-129Xe SEOP 27.
3.5 光泵的应用
医疗诊断方面,由于临床上使用的诊断方法,如肺功能测试和CT,会产生电离辐射,无法避免造成损伤,难以安全高效可行的定量检测气血交换功能等肺部生理学43。
自旋交换光泵超极化的惰性气体,尤其是129Xe气体,由于其良好的溶解度和很强的化学位移敏感性,再加上比热平衡状态下极化增强高达50000倍,近年来在肺部磁共振成像上应用广泛,能够实现对动物肺部结构和功能的检测。Lauterbur首先采用磁场梯度的方法发明了磁共振成像技术,开创了磁共振成像领域44。1994年,Albert等人在离体小鼠的肺部得到第一幅超极化惰性气体肺部磁共振影像45,自此这种方法得到了快速的发展46,47(图9)。目前,利用光泵超极化129Xe气体,已实现了活体动物的肺部通气磁共振成像48、脑部表观弛豫方法磁共振成像49,具有相当的发展前景。近年来,四极核83Kr (I = 9/2)在磁共振成像中也得到了应用50。另外,超极化氙还在研究催化剂内部结构和催化反应机理中有着广泛的应用51–53。
光泵与光检测核磁方法在砷化镓系列半导体的量子阱与异质结方面的研究也颇为广泛。例如,Kalevich等人测定了100 nm宽GaAs/Al0.3Ga0.7As量子阱的光泵核自旋极化和光检测核磁谱54;Krapf等报道了一种P掺杂Al0.36Ga0.64As/GaAs异质结的光检测核磁实验55。
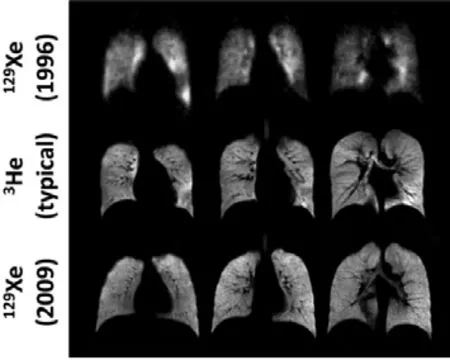
图9 利用超极化129Xe的肺部磁共振成像46Fig. 9 MRI of lung using 129Xe 46.
4 光核极化
1967年,Maier和Wolf发现蒽的单晶在非极化的白光照射下原子核会发生超极化56,后来人们发现在一些芳香族化合物的混合晶体中也能发生这种光核极化现象。这种极化现象在低磁场下产生,而且当磁场变大的时候极化依然保留57。这种极化具有较长的T1,可以把样品在低磁场下极化,然后转移到高磁场下进行测量。光核极化的过程与光泵法相似,通过光照射样品实现原子核的超极化。但是光核极化的原理与光泵法是不同的。
以芳香族化合物为例,基态时所有电子成对处于单线态S0,进行光照时会激发形成激发态单线态S1,激发态单线态会在系间窜越(Inter-System Crossing,ISC)的作用下转化为三线态T1。由于晶体中电子的自旋-轨道耦合的各项异性,三线态会裂分为三个能态(Tx、Ty、Tz零场裂分,Zero Field Splitting,ZFS)。三个能态在系间窜越时会产生不同的布居数,进而产生电子的极化(图10a)。这一过程被称作光电子极化(Optical Electron Polarization,OEP)。当施加特定的磁场时,电子的极化会在超精细耦合的作用下转移给原子核,实现原子核的超极化57。
与光泵法相比,光核极化利用晶体的各项异性,在极化电子的过程中不需要磁场和极化光。在极化转移的过程中光核极化需要外加磁场有特定的方向和大小,从而使极化转移更为高效。
含有氮-空穴(NV)中心的金刚石是很好的光核极化系统。NV中心的结构如图10b所示,金刚石中相邻的两个碳原子一个缺失另一个被氮原子取代。NV中心也被称为色心分为两种,一种是中性NV中心包含5个电子(氮原子的孤对电子,与空穴相连的三个碳原子的悬键),这种NV中心在Jahn-Teller效应的作用下对称性较差,弛豫时间短。另一种是带有一个负电荷的NV中心,其包含6个电子,电子轨道畸变小弛豫时间长58。现阶段被广泛研究的是第二种带有一个负电荷的NV中心,后面提到的NV中心也都是指这种NV中心。NV中心的基态为三线态(S = 1),在磁场的作用下可裂分为三个能态对应ms= 0, ±1。但是由于NV中心拥有C3v的对称性,在零磁场下ms= 0也会和ms= ±1发生裂分,其中ms= ±1的两个能态是简并的(图10c)59。在光激发和系间窜越的作用下ms= 0的基态的布居数会不断增大发生超极化(图10d)60。
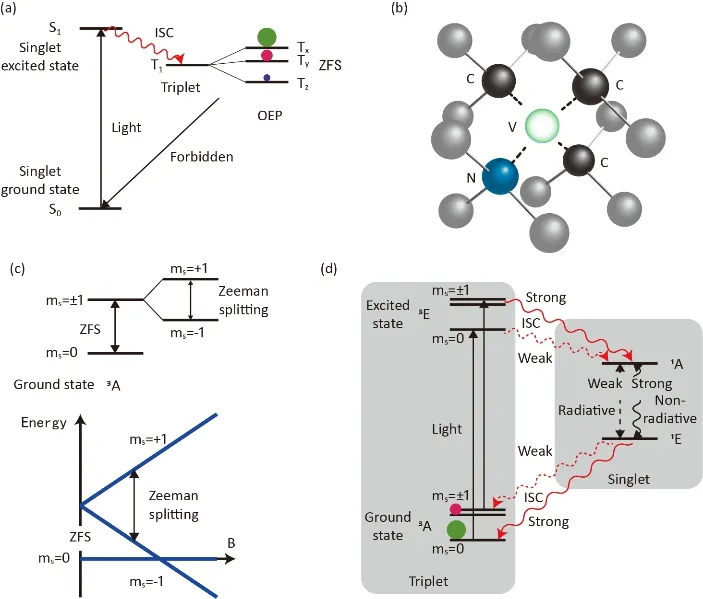
图10 (a)分子晶体的光电子极化原理示意图,其中Tx,Ty,Tz能级上面的圆形大小代表各个能级的布居数。(b)金刚石中的NV中心示意图(灰色和黑色的球代表碳原子,蓝色的求代表氮原子,绿色的空心圆代表空穴)。(c) NV中心的零场裂分和塞曼裂分。(d) NV中心的光电子极化原理示意图60Fig. 10 (a) Level diagram of molecular crystal. Circle sizes stand for the population of Tx, Ty, and Tz. (b) Scheme of nitrogen-vacancy (NV) center, grey and black balls stand for carbon, blue is nitrogen, and green is vacancy. (c) Zero field splitting (ZFS) and Zeeman splitting of NV center. (d) Scheme of optical electron polarization of NV center 60.
NV中心的极化可以通过超精细耦合转移给其附近的原子核。由于NV中心的能级裂分较大(零磁场下基态裂分为2.8 GHz),超精细耦合作用不能有效的导致电子和原子核之间的极化转移。为了实现有效的极化转移可以通过以下几种方法。第一种可以通过施加合适的外磁场使ms= ±1这两个能级发生裂分,控制其中一个态的能量与ms= 0的能量相近从而发生有效的极化转移61,62。第二种可以通过与微波照射的方法,施加一个频率与NV中心能级裂分匹配的微波并调节微波的功率使Rabi频率与原子核的Larmor频率匹配(Hartmann-Hahn条件)63–65,这样也能实现有效的极化转移。另外还可以通过固态效应,激发禁阻的双量子跃迁来完成极化转移66。上面的这几种方法对NV中心的空间取向的一致性要求比较严格,最近研究人员又提出了一种快速扫场的方法,可以实现纳米金刚石这类空间取向随机的NV中心的极化转移67。值得注意的是现阶段使用的NV中心超极化方法都是通过光检测的方法完成的。首先通过NV中心将原子核极化,极化的原子核演化一段时间后再通过微波和激光技术将核自旋态映射成电子的自旋态,最后通过检测体系的荧光来得到核自旋演化的信息68–70。
NV中心对原子核的超极化作用在未来可以被应用于单分子检测和磁共振成像中。研究人员已经实现了29Si单自旋信号的观测71。也有研究通过分析和模拟指出NV中心可以增强磁共振成像的信号72,73。
5 化学诱导动态核极化
化学诱导动态核极化(Chemical Induced Dynamic Nuclear Polarization,CIDNP)指在某些光激发或热激发化学反应中核磁共振信号发生大幅增强。该现象最早在1967年由Bargon等人74和Ward等人75分别在苯甲酰过氧化物的分解和烷基锂与卤代烷的反应中发现。1969年自由基对机理的提出完美诠释了大部分化学诱导动态核极化的现象76。这里将从CIDNP原理以及应用等方面对其进行简单介绍。
5.1 CIDNP原理
5.1.1 自由基对机理(Radical Pair Mechanism,RPM)
化学诱导动态核极化都发生在热激发或光激发化学反应中,这些化学反应都经历自由基对中间体。由于原子核对自由基电子的耦合作用,原子核自旋会影响自由基的反应活性。拥有不同核自旋的自由基会生成不同的产物,从而产生拥有极化的产物。
如图11所示77,基态反应物在光激发下形成三重态前体,只有单重态的前体能够重新结合成新的分子。在高磁场下三重态T0与单重态S在超精细耦合的作用下会发生混合,混合速率受到原子核的自旋态和两个自由基电子Larmor频率之差影响。假设原子核处于β态的自由基会更快的转变为单重态,则重新结合的反应物(或重排产物)会出现β态的富集产生发射峰。原子核处于α态的三重态在扩散作用下与其它反应物的自由基结合生成产物,出现吸收峰。
在低磁场和超精细耦合作用下,三重态T+或T−与单重态S的混合占主导。这种作用下会发生原子核自旋态会与电子态同时发生改变,三重态与单重态混合的同时,原子核的自旋态也会发生改变,从而产生极化78。
5.1.2 CIDNP线形
CIDNP主要有两种极限线形,净效应(net effects)线形和多重效应(multiplet effects)线形。净效应作用下核磁共振信号峰形和相位与普通核磁共振下得到的相同,多重效应作用下的核磁共振信号呈现正负信号交替(图12a)79。任何CIDNP的谱图都可以分解成这两种效应叠加的结果,净效应和多重效应是解释CIDNP结果最基础也是最重要的部分。
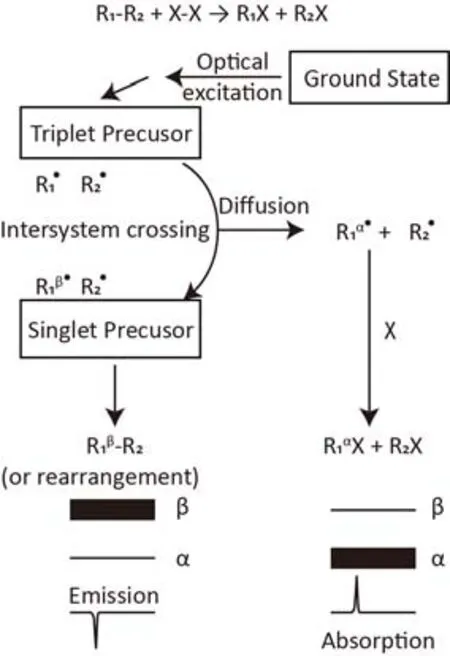
图11 CIDNP的原理77Fig. 11 Mechanism of CIDNP 77.
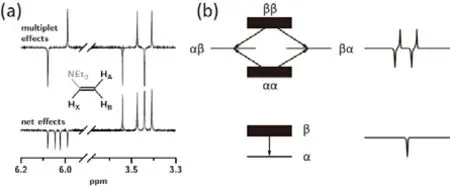
图12 (a)乙腈溶剂中激发氧蒽杂酮与三乙胺发生的光反应产生的CIDNP净效应和多重效应。图示为产物N,N-二乙基乙烯胺的谱图,上方为多重效应CIDNP;下方为净效应CIDNP。图示左半部分为HX信号,右半部分为HA和HB信号。(b)净效应(下)与多重效应(上)示意图77Fig. 12 (a) CIDNP net (bottom) and multiplet effects (top) in the photoreaction of excited xanthone with triethylamine in acetonitrile. Shown are spectra of the product N,N-diethylvinylamine (for the formula, see inset); left half, signals of HX; right half,signals of HA and HB); (b) level and population diagram of net (bottom) and multiplet (top) effects 77.
净效应中自由基对中的两个自由基电子的Larmor频率不同,只有一个原子核与自由基电子存在超精细作用。此时三重态与单重态的混合速率受两个电子频率差和超精细耦合强度影响。原子核的自旋态会在三重态和单重态的转换中富集在α态或β态,产生与普通核磁共振相同的线形。多重效应中自由基对中的两个自由基电子的频率相同,至少有两个原子核与自由基电子存在超精细作用。此时三重态与单重态的混合速率只受超精细耦合强度影响。以自由基电子与两个原子核存在超精细耦合作用为例,两个原子核的自旋态会富集在平行态(|αα〉和|ββ〉 )或反平行态(|αβ〉和|βα〉),呈现出四条正负交错的谱线(图12b)77。
CIDNP的线形可以通过Kaptein提出的相位运算规则80与产生机理相关联从而推测化学反应机理,

其中Γne表示净效应的相位符号,Γmu表示多重效应的相位顺序,根据前体多重度符号μ;产物类型ε;自由基对两个电子的频率差Δg;i核与j核的位置信息σij;i核和j核与其所在自由基的电子的超精细耦合常数的符号ai,aj;i核和j核之间的自旋-自旋偶极耦合常数的符号Jij等参数符号能计算出净效应或多重效应的相位符号。
结合极化强度和极化相位,我们可以判断产物类型进而推测反应机理。而对于一些反应过程涉及到多个自由基反应,每一个自由基反应都可能涉及到相应的核自旋筛选整理,每一对自由基都继承了上一对自由基前体的密度矩阵完整状态,可能会存在一些不易观测到的相位修正在下一步转化成极化,因此这样得到的CIDNP效应不再只是简单地根据独立自由基对寿命权重叠加,而是可能会出现比较有趣而不符合上述规律的CIDNP现象。
5.2 实验方法与脉冲序列
目前应用化学诱导动态核极化的实验方法大致可以分为两种类型:一是不含时间的CIDNP实验,二是时间分辨的CIDNP脉冲序列。两种方法都需要先对样品进行预饱和处理以消除不属于反应体系的物质背景信号(如未完全氘代的水、生物样品中加入的高浓度稳定剂等)。
对于不含时间的CIDNP实验(图13a)81,其优点在于光照时间足够长能保证激发得到的自由基数目足够多进而保证实验的灵敏度,操作简单选择性强,可以应用在任何脉冲上(包括一维和二维脉冲序列),主要用于反应中间体、反应产物以及机理的研究。它的缺点是由于光照时间过长,样品背景信号在这段时间内弛豫进而对实验造成不可避免的信号干扰。为此Goez等人通过在脉冲序列合适位置穿插180°回波脉冲使背景信号在零附近震荡并在观测脉冲作用时恰好处于零点,或通过调控在脉冲序列不同位置进行光照并设计相应相位循环来消除背景信号82。
时间分辨的CIDNP实验(图13b)81主要通过改变瞬时激光照射后的等待时间测得一系列CIDNP谱图,然后将信号归一化处理得到时间相关的CIDNP效应演化曲线来研究反应动力学过程。由于其光照时间短,光照时间范围内背景信号的弛豫可以忽略不计,然而激发的光子数目少导致相应CIDNP效应较弱,因此它面临的最重要的挑战就是提高灵敏度。除了增加扫描次数的方法,Goez等人为此设计了累计次闪光后的CIDNP效应单次采样的方法极大程度提高了谱图的灵敏度和信噪比83。

图13 (a)不含时间(静态) CIDNP简单实验脉冲示例。(b)时间分辨实验方法示例,右边为相应得到的CIDNP效应随时间演化图81。Fig. 13 (a) Principle of a CIDNP experiment without time resolution; (b) A time-resolved CIDNP experiment 81.
5.3 应用
传统核磁方法能通过一维或多维核磁共振实验得到物质的化学位移、结构信息以及动力学信息CIDNP的引入通过增强极化信号,能更明确获取痕量分子或结构组成复杂的大分子的结构信息84。特别是CIDNP极化常用于研究溶液中的蛋白质表面以及内部结构信息、活性中心85,也常用于研究有机化学反应中的反应机理研究,确证自由基反应86。如通过定量处理CIDNP谱图,可以得到自由基的超精细常数等物理参数,判断反应中间体的结构与类型,判断是否发生化学反应87,88;根据CIDNP极化的符号和相位可以得到自由基前体和产物的结构信息以及产物类型、前体多重度、自由基相关参数符号等,进而推导反应机理89;通过时间分辨的CIDNP能进一步获取与反应动力学相关的信息,如反应速率常数、电子转移速率等90,91。
6 固体中的光化学诱导动态核极化
上一章中主要讲了液体中的化学诱导动态核极化,通常由光激发或热激发,对样品分子量有较大限制,许多生物大分子由于其溶解度问题均不能通过这种方法进行信号增强进而进一步研究参与反应物质结构与反应过程,因此,发展固体条件下的化学诱导动态核极化实验(下文简称固体photo-CIDNP)成了必须。由于固体反应体系受研究对象影响大且检测条件较为严苛,直到1994年,Zysmilich和McDermott才在固态类球红杆菌R26的光反应中心首次观测到类似的化学诱导动态核极化谱图92,证明了固体下的光化学诱导动态核极化效应的存在。随后许多科学研究证明photo-CIDNP效应几乎能在所有光合作用反应活性中心被观测到;直到2010年,Thamarath等人在蓝光受体向光素LOV1-C57S突变体上发现photo-CIDNP效应93,这是首次在非光合作用体系发现固体photo-CIDNP效应。固体photo-CIDNP研究可以提供电子基态经过光循环之后的电子结构和自由基对的电子结构,对生物体系机理研究有重大意义。
6.1 固体photo-CIDNP原理
液体中的光化学动态核极化主要机理是自由基对机理,由扩散动力学驱动,不适用于固体光化学诱导动态核极化。固体photo-CIDNP可以由几种机理结合解释(图14)94,包括三自旋混合(Threespin mixing,TSM),弛豫差异(differential relaxation,DR),衰变差异(differential decay,DD),这些机理的共同点都是通过超精细相互作用将电子极化转移到核极化。
6.1.1 三自旋混合机制(Three-Spin Mixing,TSM)
光诱导电子迁移产生自旋相关的自由基对。初始产生的自由基对是单重态且高度电子极化。电子-电子-核三自旋混合体系。在自由基对存活时间内,体系由超精细相互作用的久期部分(A)、电子Larmor频率差(ΔΩ)、电子之间的偶极耦合作用(D)驱动在S和T0态之间进行零量子跃迁,同时电子自旋极化在超精细相互作用的非久期部分(B)核电子之间偶极耦合(d)驱动下转移到核极化上。为了更清晰地理解S-T0混合过程的哈密顿量变化,根据电子偶极耦合引入一个虚拟电子自旋S’ = 1/2,其两个 自 旋 态 分 别 为 |α1β2〉和 |α2β1〉。 体 系 的Hamiltonian为,
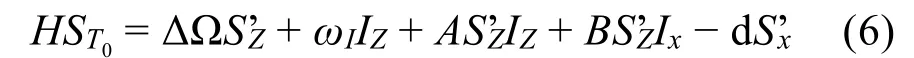
其中ωI为原子核的Larmor频率,S’i和Ii分别为虚拟电子和原子核相应方向的角动量算符。
根据各能级之间的能级差(图15)95,96可以发现当2|ΔΩ| = 2|ωI| = |A|时,其中三个能级简并,三自旋混合达到最大化,相应也使极化转移达到最大效果。然而,当电子与核之间的超精细相互作用非久期部分B为0时,电子极化转移到核极化的驱动消失,因此TSM效应消失;同样,当电子-电子偶极耦合过大时,S态和T0态趋向于形成其相应本征态,TSM效应消失95,96。
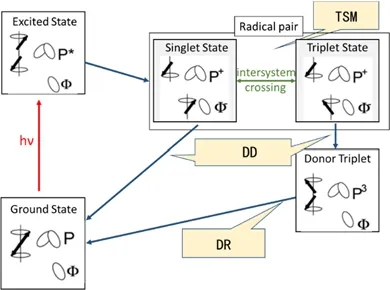
图14 产生固态光化学诱导动态核极化的三种机制94Fig. 14 Three mechanisms (TSM, DD, DR) to produce solid state photo-CIDNP effects 94.
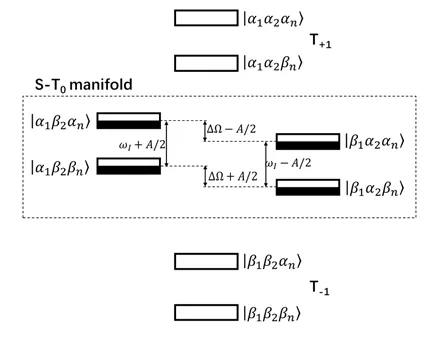
图15 三自旋混合体系能级分布图:电子的四种能级符合玻尔兹曼分布;光激发后,S态完全极化,并通过三自旋混合机制进行系间窜越达到S-T0平衡,S-T0混合的能级由于电子塞曼相互作用差异、电子-核超精细相互作用、核塞曼相互作用以及电子-电子偶极作用裂分为4个能级95,96Fig. 15 Energy levels of the radical pairs in electronelectron-nuclear three-spin mixing. The four quantum states of the S-T0 manifold are split due to the differences of the Zeeman interaction of both electron spins,the hyperfine coupling to the nuclear spin, the nuclear Zeeman interaction, and the coupling between the two electron spins 95,96.
6.1.2 衰变差异(Differential Decay,DD)
在衰变差异机理中,光化学和自旋动力学共同作用产生极化效应。TSM机理中是基于电子-电子偶极耦合作用构建了一个虚拟自旋S’,在DD中,根据三重态自由基对和单重态自由基对不同的寿命TT、TS建立虚拟自旋S’的极化,同样经由超精细相互作用的非久期部分B将电子自旋极化转移到核净极化。根据其极化机理可知,DD机理允许电子偶极相互作用d = 0,但当电子塞曼相互作用频率差或超精细相互作用的久期部分A = 0时DD效应消失。与TSM机理类似,DD机理也要求频率匹配2|ωI| = |A|,它的效率取决于由Δg和超精细相互作用驱动的自由基系间窜越时间尺度与自由基衰变时间尺度的匹配程度。当频率完全匹配,DD效率最大时,其诱发的photo-CIDNP极化与TSM机理下的photo-CIDNP极化大小相等方向相反97。
6.1.3 弛豫差异(Differential Relaxation,DR)
弛豫差异机理中,由于部分三重态衍生的极化在三联体供体存在期间发生弛豫导致三重态自由基对和单重态自由基对产生的方向相反的极化无法相互抵消进而产生净极化98。这要求特殊对三联体中核的纵向弛豫时间缩短至与三联体的寿命相当;然而由于特殊对三联体中核的纵向弛豫是未知的,因此难以预测DR机制下的CIDNP效应对总体CIDNP效应的贡献。早期很多实验观测到的photo-CIDNP效应都很难用DR机理解释;2006年Prakash课题组99发现在WT型细菌反应活性中心和R26型反应活性中心的极化模式差异可以用DR机理完美解释且后续做了许多相应研究;WT型三联体寿命100 ns,R26型三联体100 μs,如图16所示,WT型CIDNP极化相位一致而R26型部分极化相位反向,排除TSM与DD机理作用,说明DR机理在WT型反应中心CIDNP效应中的贡献明显弱于R26。这也表明核自旋弛豫时间对超精细耦合作用的依赖性与顺磁弛豫的预期效率是一致的。
6.2 固体photo-CIDNP应用
由于固体photo-CIDNP的研究起步较晚,进展较慢,早期研究大部分集中于生物体的光合作用活性中心,主要应用有如下几个方面:Photo-CIDNP效应对谱图信噪比大幅度提高有助于通过二维谱研究反应体系中痕量存在特殊自由基对的电子结构100;对生物体整个细胞甚至植物的光合系统进行结构研究101;并通过对信号弛豫的研究进一步确证生物大分子结构特性,通过研究photo-CIDNP极化的场依赖型可以得到关于自由基对寿命的相关信息,如图16所示94。同时,与液体中的CIDNP效应类似,固体中的photo-CIDNP也可以通过时间分辨的脉冲序列研究反应动力学过程,通过计算极化的强度可以得到单重态或三重态反应前驱体的信息。新发展的能将高度photo-CIDNP极化转移到邻近核的“spin-torch”实验更是有助于人们进一步对自由基对周边的环境信息进行精准研究102,如研究辅因子周围的蛋白质信息等等。固体photo-CIDNP在非光合作用体系的应用目前还只局限于向光素LOV1-C57S的蓝光受体上,发展普适性的photo-CIDNP信号增强技术还面临巨大挑战。

图16 醌耗尽的R26型(左)和WT型(右)类球红杆菌反应活性中心在不同磁场下的photo-CIDNP谱图(红色)与正常核磁谱图(黑色)。a,b,c,d从上到下分别对应磁场强度17.6、9.4、4.7、2.4 T 94Fig. 16 13C MAS NMR spectra of quinone depleted RCs of R.sphaeroides R26 (left) and WT (right) in the dark (black) and under illumination (red) at 17.6 T (a), 9.4 T (b), 4.7 T (c), and 2.4 T (d) 94.
7 仲氢诱导极化(Parahydrogen Induced Polarization,PHIP)
1981年,Bryndza在探究碳基钴炔烃络合物的加氢反应的过程中,意外地发现产物的1H NMR谱图中信号有奇妙的反相增强现象103。当时该现象被认为是化学诱导动态核极化(CIDNP)效应所引起,因此当时得到的结论是该反应体系伴随着自由基的生成。除此自外,Bryndza的研究还发现在40 K条件下,向反应系统通入氢气时间越长,信号增强效果越显著。直至1986年,Bowers和Weitekamp通过理论计算研究表明使用仲氢富集的氢气进行加氢反应,可实现NMR信号的有效增强104。他们为此极化效应命名为PASADENA(Parahydrogen And Synthesis Allow Dramatically Enhanced Nuclear Alignment),很好地解释了此前Bryndza在实验中所观测到的NMR信号增强现象。随后,PASADENA效应在实验上也被证实105,106。1988年,Pravica和Weitekamp报道了另一类NMR超极化实验。使用富集仲氢的氢气在地磁场(弱磁场)下进行加氢反应,并随后转移到NMR仪器磁体腔体内进行信号检测,可获得区别于PASADENA的信号增强效应,该类实验被称为ALTADENA(Adiabatic Longitudinal Transport After Dissociation Engenders Nuclear Alignment)107。1990年,有关仲氢的此类超极化实验被统称为仲氢诱导极化(PHIP,Parahydrogen Induced Polarization)108。
7.1 仲氢
核自旋变体是由于分子中对称位置上的同种原子的核自旋状态的不同而引变出的新分 子。氢分子有两种核自旋变体,分别为正氢(ortho-H2)和仲氢(para-H2)。对于氢气分子,当分子中两个相同的氢原子相互交换时(即位置进行反演的同时自旋互相交换),根据泡利不相容原理,分子的总的波函数要求是反对称的。而分子总的波函数的对称性只取决于转动波函数与核自旋波函数对称性的改变。因此,当核自旋波函数是对称时,对应着反对称的转动波函数(转动角量子数J为奇数),此类氢气分子称为正氢;当核自旋波函数是反对称时,对应着对称的转动波函数(转动角量子数J为偶数),此类氢气分子称为仲氢。仲氢,对应于单线态,总的核自旋量子数I = 0,自旋波函数为:

正氢,对应于三线态,总的核自旋量子数I=1,自旋波函数为:
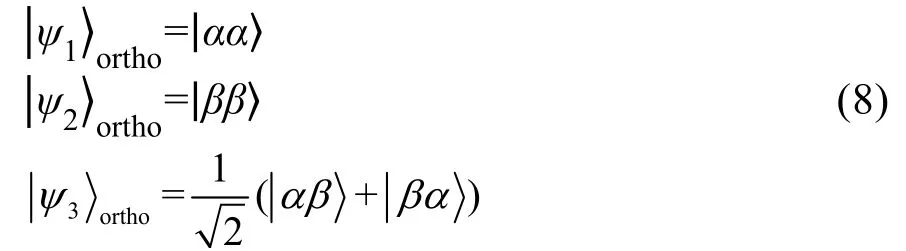
正氢-仲氢,两种自旋异构体在常温下较稳定,但随着温度的变化两者的平衡浓度会随之改变。然而,改变温度使自调节达到平衡的过程非常缓慢,通常加入催化剂可有效加速两者的相互转变。常温常压下,普通氢气中仲氢与正氢的组成之比约为1 : 3。普通氢气在经过液氮冷却并通过氧化铁催化剂催化,可获得50%仲氢富集的氢气。Schwartz等发现,在77 K下,基于低催化活性的改性沸石吸附剂可实现正氢/仲氢的高效分离,最高可实现85%仲氢的富集109。这种方法虽然也是利用沸石对正氢和仲氢吸附能力的不同而完成两个物种的分离。但是他们通过改性沸石,减少其中的顺磁缺陷(顺磁缺陷可以加速仲氢向正氢的转变)并且优化氢气通过催化剂的流速获取最大的仲氢比例。Tom等通过搭建一种新型的仲氢转换器可实现以~0.4 L∙min−1的速率连续产出丰度> 99.99%的高纯仲氢110。

图17 AX双自旋体系能级模型示意图:(a)标准NMR;(b) PASADENA;(c) ALTADENA 111Fig. 17 Level diagrams of AX spin systems: (a) standard NMR; (b) PASADENA; (c) ALTADENA 111.
7.2 PHIP的基本原理
仲氢分子(para-H2,I = 0)本身是不具有NMR信号的,NMR谱图上观察到的的氢气的1H NMR信号(通常~ 4.5 ppm)均来源于正氢(ortho-H2,I = 1)。考虑氢气分子中的两个氢核,对应双自旋系统,对于一般的NMR实验,在热平衡状态下自旋布居数在四个自旋能级(αα,αβ,βα,ββ)上的分布满足Boltzmann分布。如图17a111所示,对应可能发生的4种跃迁,反映在谱图上的是相应频率的两个双重峰(doublets)。当进行PASADENA实验时,para-H2在强磁场下进行加成反应,对于产物分子中两个来源于仲氢的氢核(AX型自旋系统),自旋布居只集中在于αβ与βα两个能级。同样地,可能发生的4种跃迁(图17b)111对应着谱图上的两个反相双重峰(antiphase doublets)。但相比于一般的NMR实验,由于自旋布居数的差异的增大,NMR信号得以提升。当进行ALTADENA实验时,para-H2在低场下进行加氢反应,并迅速转移至NMR仪器磁场下进行信号检测。在这种情况下,产物分子的自旋系统是在低场下形成的,表现出强耦合作用。此时迅速转移至高场,自旋布居只集中在αβ或βα其中一个能级。最后可能发生的跃迁只有2种,谱图上表现出相应频率的两个反相单重峰(如图17c所示)111。通过简单的自旋能级模型的分析,若要实现超极化,仲氢必须是成对低转移至产物分子中。若产物分子中的H原子分别来源于不同的H2分子,αα、ββ、αβ和βα是同等的,最后造成能级间的布居数差异减小,无法实现极化的效果。
7.3 PHIP的应用
PHIP是基于将仲氢加成至不饱和化合物上来获得NMR信号增强的一种技术,是研究加氢反应催化机理的有力手段。Yao等以乙炔的加氢反应为例,考察了氢气通入方式、反应温度和反应压强对PHIP实验中核磁共振信号增强效果的影响112。Stepanov等通过CF MAS NMR (Continuous Flow Magic Angle Spinning NMR)对Pt催化丁烯的加氢反应体系实现了PHIP效应的观测,同时对体系中的气相产物和吸附物进行直接检测,进一步地验证了Pt催化剂体系H2成对加成的反应机理113。此外,基于PHIP可有助于新型催化剂的开发。Deng等研究了Pd-Au双金属催化剂的制备方法、化学组成及结构对加氢反应活性和PHIP效率的影响114,115。
PHIP可以通过极化转移实现13C、15N等稀核的信号增强,可用于研究生物代谢反应及过程等。Goodson等通过Rh/TiO2催化剂催化神经碱的加氢反应实现极化转移15N超极化,可实现在低场下进行NMR信号检测,并发现全氘代的神经碱可有效延长PHIP效应的寿命116。Chekmenev等通过一种氘代前体的仲氢加成反应,可有效实现PHIP13C极化转移,灵敏度提升了超过107倍117。
人们也开发了可以利用PHIP信号增强的造影剂,提高磁共振成像(MRI)的检测速度和灵敏度118;此外,PHIP在低场或零场NMR、痕量分析、量子计算以及化学指纹等方面有着广泛的应用119–121。
