基于全溶体系的毛竹竹材木质素分离方法
2020-04-22吴文娟闫雪晴邹春阳王博伟
吴文娟,闫雪晴,邹春阳,王博伟,何 贤
(南京林业大学江苏省林业资源高效加工利用协同创新中心,江苏南京210037)
木质素是一种广泛分布于植物细胞壁中无定形的芳香性高聚物,是除纤维素外自然界中储量位居第2位的可再生资源。随着木质素化学分解方法研究的进展以及光谱、色谱等分析技术在木质素化学研究中的广泛应用,对木质素化学结构的认识也越加深刻。但是,人们对木质素结构的认识在很大程度上是基于对分离木质素的研究,而木质素主要分布在植物细胞的微纤丝之间,并且与高聚糖有着复杂的化学或物理链接,迄今为止尚未找到一种理想的分离方法,使木质素完全地无变化地从植物原料中分离出来[1-2],因此,分离方法在很大程度上影响着木质素的结构研究。一直以来都是用球磨木质素(MWL)来分析木质素的化学结构[3-4],但因为得率低,MWL是否能代表植物原料中全部原本木质素也存有争议[5],纤维素酶解木质素(CEL)因为得率高,且结构与MWL相似,被认为可代表植物中的原本木质素[6]。CEL的一个主要缺点为酶水解过程中的酶不能在后期分离纯化时完全除去。WU等[7]在此基础上提出了一种从阔叶木、针叶木中有效分离木质素的新方法:EMAL(enzymatic/aicdlolysis lignins)。该分离方法结合酶处理与温和酸水解,使木质素-碳水化合物间的化学链接能在温和的化学条件下,选择性地有效打开,为木质素高效分离提供了保证。相对于传统的球磨木质素,该分离方法通过球磨、酶水解及温和酸水解的协同作用,可以明显提高木质素的得率。但众所周知,酸处理将导致木质素α-醚键的断裂。LEE等[1]和MESHITSUKA等[8]研究表明:球磨后木质纤维原料木质素在二氧六环-水溶液中的溶出与球磨时间有关,胞间层木质素较易溶出,随着球磨时间的延长,次生壁木质素的溶出逐渐增多。因此,利用传统方法分离木质素,缩短球磨时间不仅降低了分离木质素的得率,也使得到的木质素缺乏代表性。木质纤维原料由纤维素、半纤维素和木质素构成,纤维素大部分分子规律性排列形成结晶结构,只有部分分子规律性差,形成无定形区。纤维素酶只能作用于植物原料的非结晶区,对于结晶区并不起作用,如果直接将木质纤维素用于酶解,效果很差。因此,在酶解前必须先改变纤维素的晶体结构,才能促进纤维素的降解,达到提高酶解效率的作用。选用合适溶剂进行溶解再生来改善结晶区的结构可以提高酶解效率[9-10]。WANG等[11]提出的LiCl/DMSO全溶体系,作用于球磨木粉,结果表明:再生前后的木质素结构单元没有发生变化,而纤维素的结晶区受到一定程度的破坏。本研究以毛竹Phyllostachys edulis竹材为原料,借助LiCl/DMSO溶剂体系溶解再生处理,通过纤维素酶水解分离出非木材木质纤维原料中的木质素,并与传统方法得到的MWL和CEL进行了比较。
1 材料与方法
1.1 原料
试验毛竹竹材产自浙江安吉,人工劈成长3~4 cm,宽、厚 2~4 mm的小竹条。上述风干原料用Wiley微型粉碎机粉碎,取40~80目组分,用苯醇(2∶1,V/V)抽提8 h,经真空干燥后,储存于带磨砂玻璃塞的广口瓶中,供分析使用。
1.2 球磨
竹粉的球磨在德国Fritsch微型行星式高能球磨机中进行。称取2 g真空干燥后的脱脂竹粉装入容积为45 mL氧化锆制的罐子,内装18只内径1 cm的氧化锆球,以600 r·min-1进行不同时间的球磨。球磨在冷库(-20℃)进行,每运行15 min,休停5 min,以避免过热。磨后竹粉经干燥后,备用。
1.3 LiCl/DMSO溶解和再生
球磨2 h的竹粉以质量分数7.5%溶于8%LiCl/DMSO溶剂体系,室温下搅拌48 h,在60℃继续搅拌24 h。LiCl/DMSO处理后,将溶解物置入透析袋,放入装有去离子水容器中,用新鲜去离子水更换容器中透析出来的水数次,直至用硝酸银无法检测出透析液中氯离子(Cl-)。再生的原料经冷冻干燥后,得到再生原料,再生得率为86.0%,高聚糖的得率81.0%。再生原料室温保存,作酶水解备用。
1.4 木质素的分离
竹材MWL的分离方法参照文献[3-4]。球磨2 h的竹粉10 g,用96%二氧六环/水(V/V)室温下提取24 h,离心分离后,残渣继续用96%二氧六环/水(V/V)体系提取1次,离心分离残渣。收集2次离心后的上清液,在30℃下旋转蒸发浓缩至干,在室温下真空干燥后,溶于体积分数为90%醋酸溶液中,离心分离,上清液缓缓滴加至10倍体积的水中,沉淀下来的固体残渣用水离心洗涤3次后真空干燥。干燥后的木质素溶于1,2-二氯乙烷/乙醇(2∶1,V/V)的混合液中,离心后,上清液于大量乙醚中沉淀,沉淀物用乙醚洗3次,五氧化二磷真空烘干箱干燥,得到竹材MWL,得率为5.1%(基于竹材Klason木质素)。
竹材CEL的分离方法参照文献[6]。取球磨竹粉和再生球磨竹粉各10 g于 500 mL锥形瓶中,加入一定量纤维素酶酶活(以滤纸酶活计)为30 μmol·min-1·mL-1的混合酶液(Novozymes提供),以每克绝干葡聚糖为基准,酶用量均为30 μmol·min-1,补充一定量的pH 4.8的醋酸-醋酸钠缓冲液,调节酶水解底物质量分数为 5%,将锥形瓶置于恒温振荡器(SHA-C)中,在 180 r·min-1,50℃下振荡72 h。水解后在5 000 r·min-1下离心15 min,分离酶水解液。残渣用蒸馏水离心洗涤3次后,经冷冻干燥和五氧化二磷真空干燥后,用96%二氧六环/水(V/V)提取,得到粗竹材CEL和再生酶解木质素(RCEL)。经上述纯化步骤后,得到竹材CEL和RCEL,得率分别为25.3%和48.2%(基于竹材Klason木质素)。
1.5 木质素的乙酰化反应
木质素的乙酰化参考文献[12]。称取300 mg木质素样品于50 mL三角烧瓶中,加入15 mL吡啶/乙酸酐(1∶1,V/V),盖紧瓶塞,搅拌,室温下反应24 h。反应结束后,用无水乙醇将反应体系蒸发至干,至无吡啶气味。加入1 mL氯仿完全溶解,并滴入100 mL乙醚中沉淀出乙酰化木质素,用乙醚洗涤沉淀多次后,置入40℃真空干燥箱中干燥。乙酰化木质素用于凝胶渗透色谱(GPC)和核磁共振氢谱(1H NMR)的测定。
1.6 分析方法
1.6.1 化学成分测定 木质素含量、灰分的含量按照文献[13]的方法测定。称取样品0.300 0 g,加入3 mL质量分数为 72%硫酸,在18~20℃下水解3 h。期间定时搅拌,然后转入 100 mL玻璃瓶中,加水将硫酸稀释至质量分数为 4%,密封后置于灭菌锅(DSX-280B)内,在 121℃下水解 1.5 h。用已恒质量的G3砂芯漏斗分离残渣和水解液,收集滤液测定酸溶木质素和高聚糖质量分数。残渣用热蒸馏水洗涤至滤液,用质量分数为10%氯化钡检测无白色沉淀,在恒温干燥箱中于 105℃下恒量,称量后转入马弗炉中于575℃灼烧至恒量,测定木质素中的灰分质量分数。计算 Klason木质素质量分数时扣除灰分的质量分数。以质量分数为4%硫酸为参比,用日本岛津公司的UV-240紫外-可见分光光度计测定水解液在波长205 nm处的吸光度,计算酸溶木质素的质量分数。
还原糖的测定参照文献[14]。取水解液5 mL,加入内标1 mL肌醇溶液(质量浓度为1 g·L-1),用饱和氢氧化钡溶液调节pH至5.5,离心分离除去沉淀,加入20 mg硼氢化钠于清液中,静置24 h。加入1 mL醋酸后,旋转蒸发至浆状,再加入甲醇继续蒸发至干,1 mL·次-1,连续3次。将蒸干体系于105℃烘干15 min以确保体系完全无水。加入1 mL乙酸酐后密封,在120℃下反应3 h。用日本岛津公司的GC-14b气相色谱仪分析还原糖。气相色谱条件如下:毛细管柱TC-17(0.25 mm×30 m);FID检测器;柱温程序,220℃保留20 min。进样温度为220℃,检测温度为230℃;气相色谱GC分析得到的单糖均转换为高聚糖。同时进行2次测定,取其平均值。
1.6.2 硝基苯氧化 木质素的结构变化可以通过硝基苯氧化结果来分析。硝基苯氧化操作参照文献[15]。取样品40 mg于10 mL不锈钢制的罐子里,依次加入硝基苯0.25 mL,2.0 mol·L-1氢氧化钠4 mL。将上述混合体系密封置于170℃油浴中反应2 h。冷却至室温,加入1 mL 0.1 mol·L-1氢氧化钠配制的内标3-乙氧基-4-羟基苯甲醛(0.2~0.4 g·L-1)。 用二氯甲烷萃取 3次, 15 mL·次-1, 弃油相取水相, 用 4.0 mol·L-1盐酸调节pH至1.0,依次用二氯甲烷萃取2次,20 mL·次-1,乙醚1次,15 mL。收集有机相共55 mL,用去离子水20 mL洗涤有机相,经无水硫酸钠干燥,过滤,滤液蒸发至干,加入100 μL N,O-双(三甲基硅)乙酰胺衍生化试剂(BSA)于100℃反应10 min。产物用日本岛津公司的GC-17A气相色谱仪进行分析。气相色谱条件如下:毛细管柱NB-1(0.25 mm×30 m);FID检测器;柱温程序,150℃保留15 min后,以3℃·min-1升温到180℃,继而以10℃·min-1升温到280℃,保留5 min。进样温度为250℃,检测温度为280℃。同时进行2次测定,取其平均值。
1.6.3 元素分析 用德国Elementar公司的ELⅢ型元素分析仪来检测样品中规定碳、氢、氮和氧的质量分数。
1.6.4 热重分析 采用Henven公司的热重分析仪进行木质素样品热失重分析,实验温度为30~900℃,升温速率为20℃·min-1,高纯氮气保护。
1.6.5 红外光谱 采用KBr压片法在日本JASCO IR-615型红外光谱仪上测定样品,波长范围为4 000~400 cm-1。
1.6.6 凝胶渗透色谱(GPC)分析 用美国Waters公司 1515型凝胶渗透色谱分析系统来测定木质素的分子量和分子量分布。GPC分析在Waters 2414折射只是检测器上进行,采用StyragelHR1, HR3和HR4色谱柱(7.8 mm×300.0 mm),测定质量浓度5~10 g·L-1,用四氢呋喃作为溶剂,溶剂流速在30℃为1.0 mL·min-1。以标准聚苯乙烯(分子量范围为100~500 000)作为标准样品制备标准曲线。
1.6.7 X射线衍射 纤维结晶指数分析在日本理学UltimaⅣ组合型多功能水平X射线衍射仪上进行,采用粉末法。测试条件:X光管为CuKα靶(λ=0.154 18 nm),用石墨单色器消除CuKα辐射,管电压40 kV,管电流40 mA,测量方法采用θ/2θ联动扫描,索拉狭缝为0.04 rad,接收狭缝0.15 mm,发射和防散射狭缝 1°, 扫描范围为 2θ=5°~40°, 扫描步长为 0.02°, 扫描速率为 5°·min-1, 记录 “衍射强度-2θ”曲线。结晶指数可以根据Segal公式[16]进行计算:
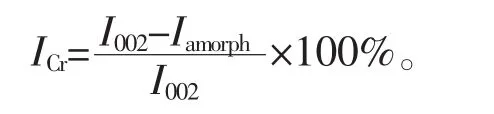
其中:ICr为结晶指数,Iamorph为 2θ为18°时无定形区衍射峰的强度,I002为主结晶峰 002的最大衍射强度。
1.6.81H NMR测定 将乙酰化木质素溶于氘代氯仿(CDCl3),在日本JEOL公司的Alpha 500型核磁共振仪上进行测样。以三甲基硅烷作为内标。
2 结果与分析
2.1 竹材木质素的特性
2.1.1 竹材CEL和RCEL的得率、化学组成 相对于传统的木质素分离技术,由于部分木质素和碳水化合物间存在着共价键链接[17],又有部分的木质素-碳水化合物复合体(LCC)溶于二氧六环与水的溶液中[18-19],使得从纤维原料中分离得到高纯度、高得率木质素受到一定局限。经比较分析,利用LiCl/DMSO溶剂体系对球磨竹粉的溶解再生可高效分离木质素。对竹材来说,球磨2 h再经LiCl/DMSO溶剂体系的溶解再生分离得到的木质素得率可达48.2%,是CEL方法的2倍多,是MWL方法(得率为5.1%)的9倍。球磨时间的缩短,木质素的溶出可借助于LiCl/DMSO溶剂体系对物料的渗透润胀来改善,使之有更多的木质素裸露出来,通过聚糖的酶解来提高木质素在有机溶剂中的溶出,这样可以避免因为长时间的球磨带来木质素结构上的损伤。所以LiCl/DMSO溶剂体系的处理有助于提高竹材木质素的得率。另外,从表1可看出:竹材的RCEL总糖质量分数为9.0 g·kg-1,CEL为 12.0 g·kg-1。RCEL的纯度略高于CEL。
由表2可以看出:碳、氢和氧元素质量分数差别不大,竹材CEL和RCEL的氮质量分数均为3 g·kg-1,氮元素在木质素中质量分数均在10 g·kg-1以下,不会影响到其化学结构的分析[20]。

表1 竹材CEL和RCEL的得率和纯度Table 1 Isolation yield and sugar content of purified lignin samples from bamboo

表2 竹材CEL和RCEL的元素分析Table 2 Element analysis of CEL and RCEL isolated from bamboo
2.1.2 分子量分析 凝胶渗透色谱(GPC)是一种木质素分子量快速测定的简单可靠的方法。但是木质素是不均一的高聚物,分析其不均匀性,即多分散性对了解整个木质素分子量的变化有重要影响。由表3可以看出:竹材的CEL和 RCEL的重均分子量(Mn)和数均分子量(Mw)相近,数均分子量约6 000,重均分子量12 000。RCEL的得率高于CEL。LiCl/DMSO溶剂体系的溶解润胀,一方面破坏了部分与木质素相连的纤维素结晶区,纤维素被酶水解成单糖溶出,木质素则留在底物里;另一方面,通过破坏纤维素结晶区,会将原来彼此包覆的木质素释放出来。最终都会导致分离木质素得率的增加。从表3看到:RCEL的重均分子量高、分散性大,说明非木材纤维原料的木质素大分子之间仍存在着较大的差异,体现了非木材纤维原料木质素大分子结构的复杂性。竹材RCEL的分散性与CEL一致,都为2.0。

表3 竹材CEL和RCEL分子量Table 3 Average molecular weight and polydispersity index (Mw/Mn) of CEL and RCEL from bamboo
2.1.3 红外光谱分析 红外光谱能反映木质素的微细结构,根据木质素红外光谱特征吸收谱带测定分析木质素所带有的功能基,例如羰基、羟基、甲氧基、C—H键和C—C键等[21]。由图1可看出:竹材CEL和RCEL红外光谱特征峰基本一致,表明两者的官能团组成基本相同,CEL和RCEL在其结构上没有明显的差异:表征苯环骨架伸缩振动的1 597、1 512和1 419 cm-1吸收峰均显示出较强的吸收,这是木质素结构的基本特征峰,说明其苯环骨架结构保存完好。在1 704 cm-1出现明显吸收峰,这些吸收峰是非共轭羰基、酯基的特征吸收峰;另外,在1 327 cm-1有紫丁香基的吸收,愈创木基单元的伸缩振动在1 234 cm-1也有明显吸收,表明CEL和RCEL中均含有一定量的愈创木基单元、紫丁香基单元。木质素芳环单元结构在LiCl/DMSO溶解再生过程中没有明显破坏。对吸收峰归属的划分结果见表4。
2.1.4 X射线衍射分析 图2为原料竹材、球磨竹材及溶解后再生竹材的X射线衍射曲线,未经任何处理的原料竹粉(B)结晶度(ICr)为58.4%,再生竹材(RMB)与球磨竹材(MB)ICr分别为26.5%和24.6%。由图2可以看出:竹材的曲线明显不同于球磨竹材和再生竹材。结合表5的结晶度可以看出:对于溶解后再生的球磨竹材原料,其得率为86.0%,高聚糖的得率81.0%,即处于无定形区的部分碳水化合物会在LiCl/DMSO处理过程中溶出,因而其结晶度略高于球磨样品。与40~80目的原料相比,结晶区有了明显的破坏。对于经过溶解再生后的样品,一方面可改善聚糖的酶水解,另一方面溶剂体系的深入渗透,会有更多的木质素敞露出来,有利于后续有机溶剂的提取、分离。
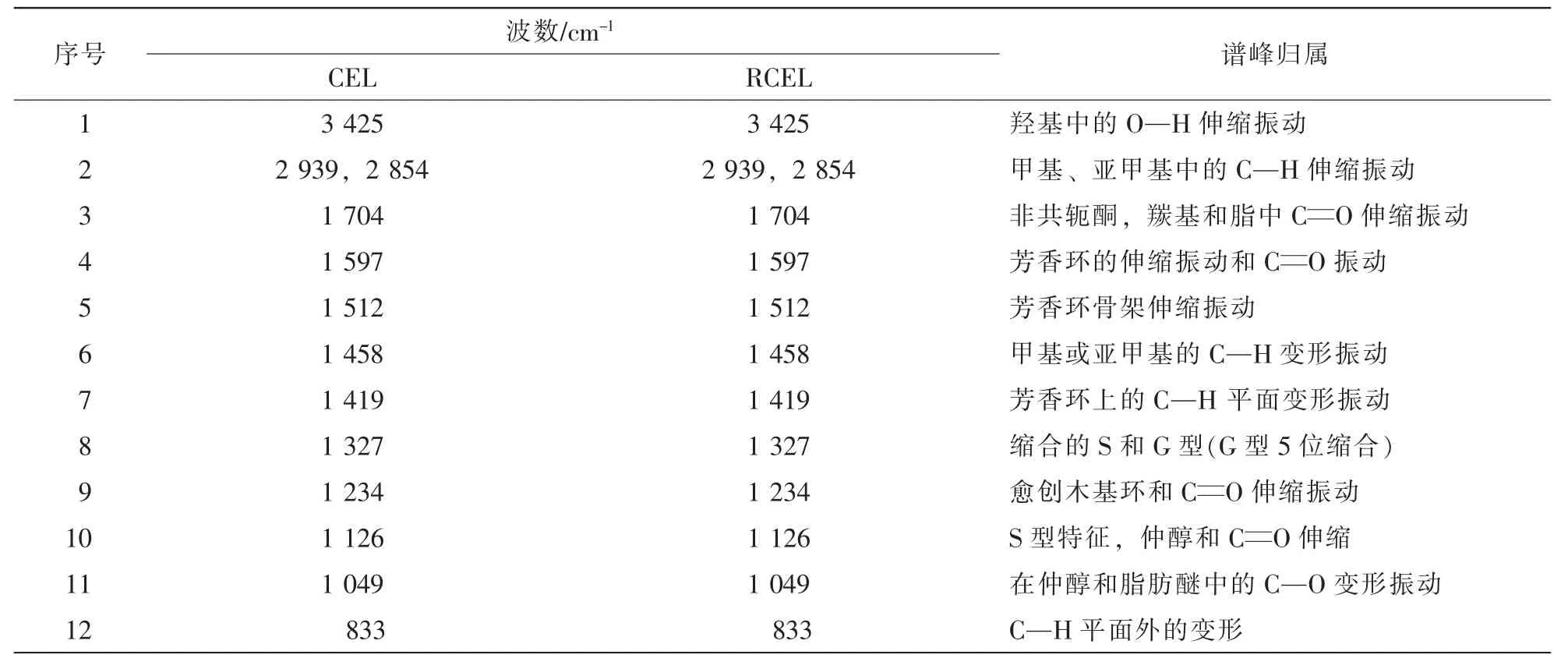
表4 竹材CEL和RCEL的红外光谱图解析Table 4 Assignment of FTIR spectra of CEL and RCEL isolated from bamboo

图1 竹材CEL和RCEL的红外谱图Figure 1 FT-IR spectra of CEL and RCEL isolated from bamboo
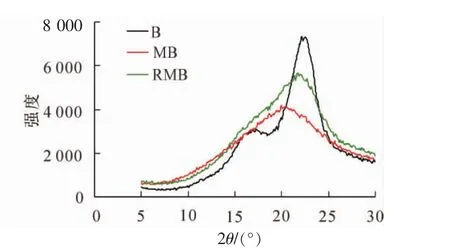
图2 竹材(B)、球磨竹材(MB)及再生竹材(RMB)的X射线衍射图Figure 2 X-ray pattern of original bamboo (B),ball milled bamboo (MB),and regenerated bamboo with ball milling (RMB)
2.1.5 木质素结构单元 碱性硝基苯氧化一般用于木质素的结构单元分析,未缩合的对羟基苯基(H)、愈创木基(G)和紫丁香基单元(S)在碱性高温条件下分别氧化为对羟基苯甲醛、香草醛和紫丁香醛。木质素结构中未缩合单元含量越高,则木质素的缩合程度越低。表5显示了酶解木质素和溶解再生后的酶解木质素的碱性硝基苯氧化结果。从表5可以看出:竹材CEL的得率为2.7%,RCEL为2.5%,CEL的未缩合程度要高于RCEL。另外,硝基苯氧化的结果表明:竹材CEL和RCEL均属于GSH类型的木质素,含有丰富的紫丁香基单元,其中紫丁香基单元在未缩合单元中的占比达一半。从对羟基苯基、愈创木基和紫丁香基单元的得率、比例及紫丁香基单元的质量分数可说明2种分离出来的木质素的结构单元比例没有明显的不同。

表5 竹材CEL和RCEL的硝基苯氧化产物的得率及S/GTable 5 Nitrobenzene oxidation products yields and S/G molar ratio of CEL and RCEL isolated from bamboo
2.1.6 木质素的热稳定性 木质素是一种复杂、非结晶性的空间立体结构的大分子有机物,以苯基丙烷单元为主体,经各种醚键、碳碳键等连接而成,且含有丰富的羟基和甲氧基等官能团,结构的复杂性会影响其热裂解[22]。由图3可以看出:竹材CEL和RCEL的热解曲线在100~800℃的热重曲线大致相同。在200~600℃内,残余物质量分数由94.4%减少至33.2%,减少了63.9%。超过600℃时,木质素中热解残余物中已不存在木质素苯丙烷单元,此时的苯丙烷单元组成的大分子结构发生了彻底的解构[23]。在600~800℃内RCEL残余物质量分数的减少值为5.0%,CEL为7.0%,此热解特性的差异主要在于木质素中化学结构的不同。经前述分析,竹材RCEL的缩合程度要高于CEL,从而导致其热解需要更高的能量。分析结果表明:竹材CEL和RCEL的热分解过程主要发生在200~600 ℃。
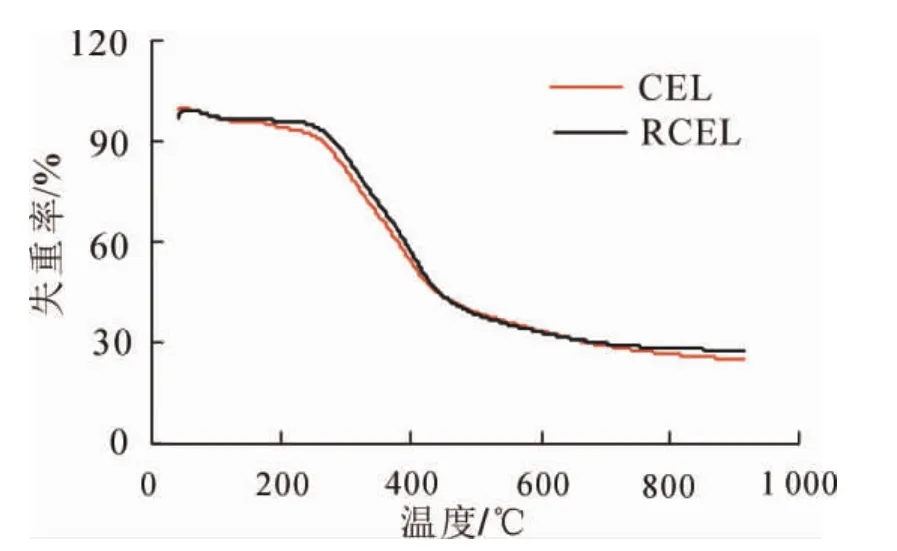
图3 竹材CEL和RCEL的TG曲线Figure 3 Thermogravimetric analyses of CEL and RCEL of bamboo
2.1.71H NMR分析 由图4可看出:竹材CEL和RCEL在结构单元方面没有任何差异,化学位移在6.25×10-6~6.80×10-6,6.80×10-6~7.20×10-6及 7.30×10-6~7.60×10-6分别对应于紫丁香基、愈创木基及对羟基苯基单元,分离出来的CEL和RCEL都属于GSH型木质素。化学位移处在3.48×10-6~4.00×10-6有较强烈的谱峰吸收,应归属于甲氧基上质子。从图4发现:2种分离木质素CEL和RCEL的各种官能团包括羟基和甲氧基峰形都十分明显,表现出丰富的官能团特征,表明分离木质素的官能团保持较好。另外,化学位移在 1.60×10-6~2.22×10-6内为芳基侧链脂肪族醋酸酯上质子,2.22×10-6~2.50×10-6的谱峰归于芳香族醋酸酯上质子。

图4 竹材CEL和RCEL的1H NMR谱图Figure 4 1H NMR spectra of CEL and RCEL of bamboo
3 结论
以竹材为原料,经球磨、LiCl/DMSO溶剂体系溶解、再生处理、酶水解得到了RCEL。与传统的方法相比,RCEL得率和纯度都较高。经表征,此分离方法对木质素大分子结构有较好的保护作用,对碳水化合物有很好的降解作用,RCEL能较好地代表竹材的木质素。
分离得到的竹材木质素为GSH型木质素,缩合程度略高,竹材RCEL中紫丁香基结构单元含量较高,并占到一半。从分子量分布来看,竹材RCEL数均分子量和重均分子量都较高,分离过程中木质素降解较少。热解特性曲线分析表明:热失重温度为200~600℃,600℃后失重趋于稳定。在800℃,竹材RCEL未被分解的质量分数为241 g·kg-1。木质素缩合程度的不同决定了其热解特性的差异。
