HPLC法测定兽用除虫脲原料药含量及有关杂质
2020-04-17左三龄邱银生吴仲元
左三龄,张 博,邱银生,叶 纯,吴仲元
(武汉轻工大学动物科学与营养工程学院,武汉 430023)
目前国内外猪场普遍存在苍蝇污染问题,养殖场蝇蛆的控制是困扰养殖生产的一大难题。控制策略主要是使用敌敌畏和除虫菊喷雾剂,但毒副作用较大[1]。除虫脲(diflubenzuron),化学名1-(4-氯苯基)-3-(2,6-二氟苯甲酰基)脲(图1)。属苯甲酰脲类杀虫剂,作用性质为昆虫生长调节剂,能够阻碍昆虫表皮中几丁质的合成,使昆虫变态受阻不能蜕皮而死亡[2-3]。兽用除虫脲作用方式主要为胃毒和触杀,无内吸收特性[4]。除虫脲的合成路线有多种[5-7],主要合成工艺为2,6-二氟苯甲酰胺与对氯苯基异氰酸酯在二甲苯溶液中缩合生成除虫脲。该农药于1976年,经美国环保局批准投入批量生产[8],2016年1月美国Central Life Sciences公司已开发出0.04%和0.67%的兽用除虫脲预混剂Mix Clarifly®作为猪、牛和马的杀蝇蛆剂使用,但目前国内无相关兽药制剂,且国内外对其原料药含量测定和有关物质检查少有相关报道。为完善兽用除虫脲的质量控制,本研究拟通过使用高效液相色谱(HPLC)建立兽用除虫脲含量测定及其有关物质检查的方法,以期为国内外进一步的兽用制剂质量标准的控制研究提供参考。
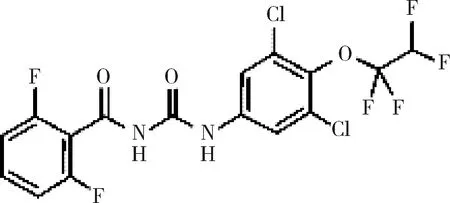
图1 除虫脲结构式
1 仪器与试剂
高效液相色谱仪(ACQUITY Arc HPLC)配备二极管阵列(PDA)检测器,购自美国Waters公司;精密分析天平(BS 110S),购自北京赛多利斯仪器系统有限公司;超纯水仪购自德国 Merck-Millipore公司;涡旋混合仪(FW80微型)购自上海沪西分析仪器厂有限公司;pH计(STARTER 2100)购自奥豪斯仪器上海有限公司;超声仪(KQ5200)购自昆山市超声仪器有限公司;药物稳定性检查仪(WD-A)购自天津天光光学仪器有限公司。
除虫脲对照品(批号G139525,含量≥99.2%)购自Dr. Ehrenstorfer GmbH公司;3批兽用除虫脲原料药(自制,批号2018090201、2018090202、2018090203);除虫脲可能的杂质(表1):A、B、C、D(含量均大于98.0%)购自MACKLIN公司;甲醇、乙腈(色谱纯)购自Fisher Cheical公司;乙酸铵、磷酸、盐酸、氢氧化钠(分析纯)购自国药集团化学试剂有限公司。
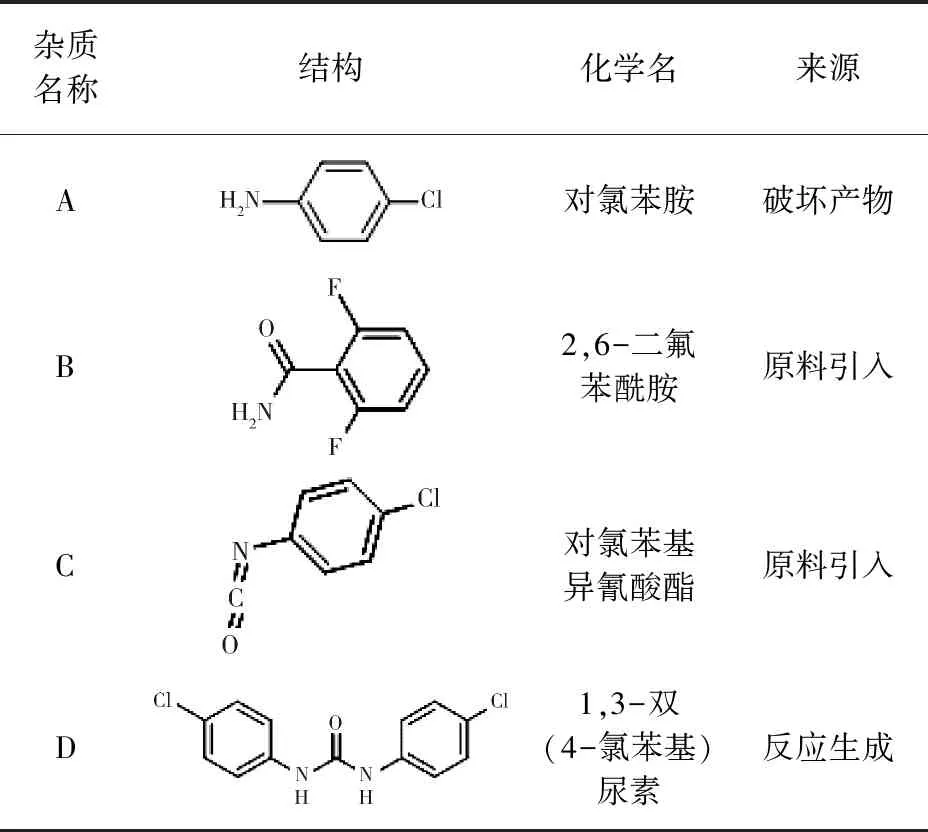
表1 除虫脲可能的杂质
2 方法与结果
2.1 含量测定方法与结果
2.1.1 色谱条件 色谱柱:Diamonsil C18(250×4.6 mm,5 μm),检测波长为254 nm,流速为1 mL/min,柱温为25 ℃,进样量为10 μL,以甲醇为流动相A,0.01 mol/L乙酸铵溶液为流动相B,进行梯度洗脱,洗脱顺序为0 min,40% A;0~5 min,40% A;5~20 min,40%~80% A;20~30 min,80% A;30~35 min,80%~40% A;35~40 min,40% A。
2.1.2 溶液配制 除虫脲储备液的制备:分别精密称量除虫脲对照品和原料药100 mg,加甲醇溶解,制成浓度为1000 μg/mL的除虫脲对照品和原料药储备溶液,4 ℃冷藏备用。
2.1.3 系统适用性试验 将2.1.2项除虫脲对照品和原料药的储备液,分别用甲醇稀释为100 μg/mL,分别进样,记录色谱图,结果见图2。计算得到的主峰理论塔板数均大于25000,各组分的分离度均大于1.5,符合规定。
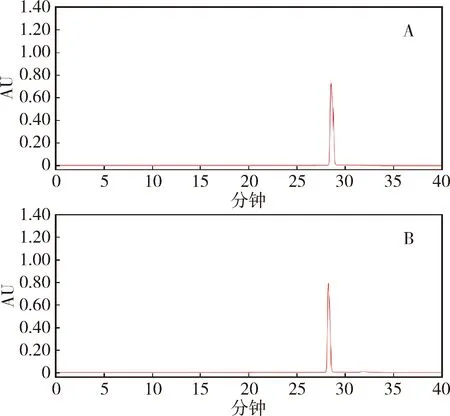
图2 除虫脲对照品(A)和原料药(B)的HPLC图谱
2.1.4 线性关系考察 将2.1.2项除虫脲对照品溶液,用甲醇分别稀释为10、20、50、100、200、500 μg/mL,以质量浓度(C,μg/mL)为横坐标,峰面积(Y)为纵坐标作线性回归,得线性回归方程为y=28 812C-1 957.7,R2=0.999。表明除虫脲质量浓度在10~500 μg/mL范围内线性关系良好。
2.1.5 重复性、精密度和稳定性试验 根据《中华人民共和国兽药典(2015版)》一部的药品质量标准分析方法指导原则,精密称取6份10 mg的除虫脲对照品,用甲醇溶解定容至100 mL,连续进样6次,RSD=0.45%,表明本方法重复性良好;将质量浓度为100 μg/mL的除虫脲对照品溶液重复进样9次,计算可得其RSD=0.13%,表明本方法精密度良好;又分别于0、2、4、8、12、24 h进样记录峰面积,用外标法计算不同时间点相对于0 h时的除虫脲的降解率为0.51%,RSD=0.20%,表明在常温条件下,24 h内除虫脲原料药溶液稳定性良好。
2.1.6 回收率试验 加入除虫脲标准品,用甲醇分别配制80%、100%、120%的供试溶液,每个3份,用外标法计算平均回收率及RSD。回收率为97.01%~100.83%,RSD<1.0%(n=3),符合回收率要求。
2.1.7 含量测定 取三批除虫脲原料药配制供试品溶液和对照品溶液进样测定,并记录峰面积,用外标法计算除虫脲的含量。三批除虫脲原料药的含量分别为99.73%(2018090201)、99.61%(2018090202)、99.67%(2018090203)。
2.2 有关物质测定方法与结果
2.2.1 色谱条件 同2.1.1项色谱条件。
2.2.2 溶液配制 据文献[9]及除虫脲合成原料及工艺中发现可能的杂质有A、B、C、D。其中C(对氯苯基异氰酸酯)需进行衍生化处理成在流动相中稳定的物质,再进样检测。根据文献[10-12]衍生化方法,C与甲醇进行酯化反应。
供试品溶液:称取除虫脲及其有关物质A、B、C、D各0.05 g,将C衍生化处理后与A、B、D混合于100 mL量瓶中,加甲醇定容,制成质量浓度均为500 μg/mL的混合对照品溶液。
2.2.3 系统适用性试验 将2.2.2项混合对照品溶液,按2.1.1项色谱条件测定,色谱图见图3。计算得到的理论塔板数均大于25000,各组分的分离度良好,均大于1.5,符合规定。
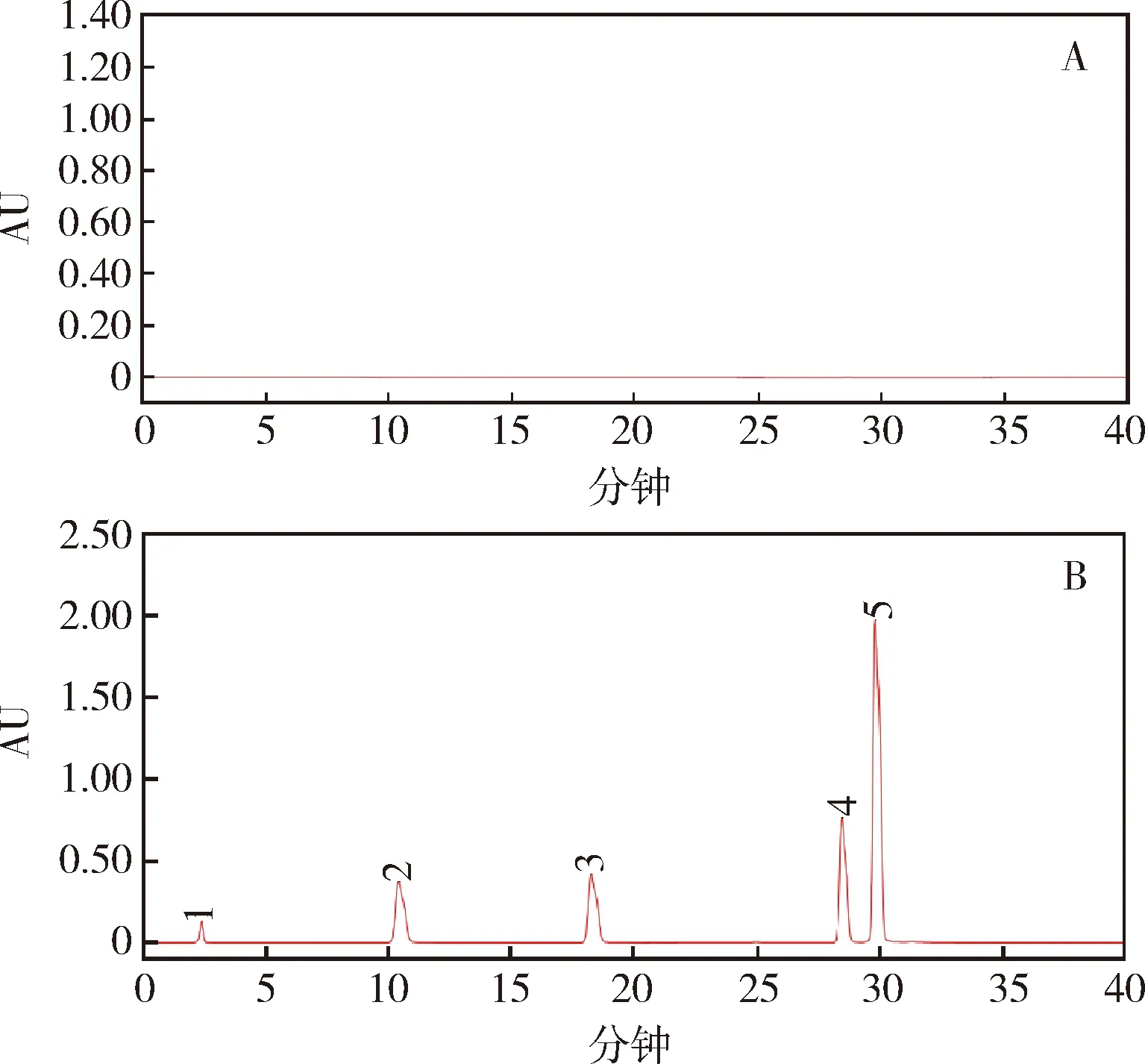
1:2,6-二氟苯酰胺;2:对氯苯胺;3:对氯苯基异氰酸酯;4:除虫脲;5:1,3-双(4-氯苯基)尿素
2.2.4 专属性与破坏性试验 分别精密称取5份除虫脲原料药0.05 g于100 mL量瓶中,处理如下:①氧化破坏:加入30%过氧化氢1 mL,超声20 min;②酸破坏:加入1 mol/L盐酸溶液(溶剂甲醇)70 mL,90 ℃水浴加热100 min;③碱破坏:加入1 mol/L氢氧化钠溶液70 mL,90 ℃水浴加热100 min;④高温破坏:于105 ℃放置10 h;⑤光照破坏:室温下,5 000±500 Lux强光照放置14 h,自然光照放置10 d。将未经降解试验的原料药和降解破坏样品加甲醇,完全溶解后,酸碱破坏样品分别中和后甲醇定容,进样测定。专属性和破坏试验的结果见图4和表2。可见未经破坏除虫脲原料药的相对保留时间(Rt)为28.437 min,氧化破坏则主要产生了降解物R1(Rt为1.404 min),酸碱、高温和自然光破坏条件下产生了降解物R2(Rt为10.411 min),强光和自然光条件下产生了降解物R3(Rt为29.624 min),强光下主要产生了降解物R4(Rt为31.802 min)。根据PDA扫描光谱图,可知除虫脲主峰和主要降解物R1、R2、R3、R4均为单一纯度峰。各降解产物与主峰的分离度R均大于1.5,符合要求,说明该方法良好。

A.空白溶剂;B.未经破坏原料药;C.氧化破坏;D.强酸破坏;E.强碱破坏 F.高温破坏;G.强光破坏;H.自然光破坏A.Blank solvent;B.Untreated raw material drug;C.Destroyed by oxidation;D.Destroyed by acid;E.Destroyed by alkali;F.Destroyed by heat;G.Destroyed by strong light;H.Destroyed by natural light
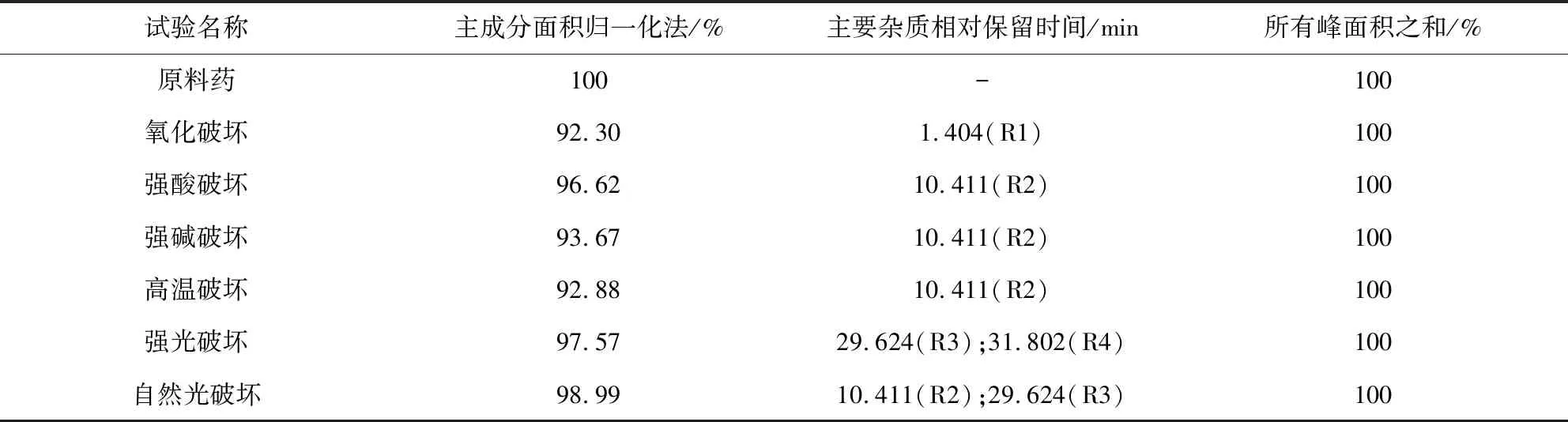
表2 破坏试验的有关物质
其中对氯苯胺与强碱、强酸、高温和自然光破坏产生的降解物R2的PDA扫描光谱图见图5,对比后发现,R2与对氯苯胺的出峰时间和最佳波长几乎一致,有理由怀疑R2为对氯苯胺。PDA扫描图谱的对比分析为除虫脲有关物质的定性检测提供了参考依据。
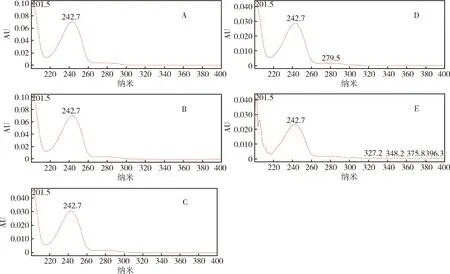
A.对氯苯胺;B. 强碱破坏;C. 强酸破坏;D. 高温破坏;E. 自然光破坏A.4-Chlorophenyamine;B. Destroyed by alkali;C. Destroyed by acid;D. Destroyed by heat;E. Destroyed by natural light
2.2.5 杂质检测限与定量限 将2.2.2项混合对照溶液,用甲醇逐步稀释后进样测定,以信噪比S/N=3、S/N=10测定,杂质A、B、C、D检测限分别为0.5244、1.5261、0.4691、0.0997 μg/mL(S/N=3),定量限分别为1.7479、5.0869、1.5638、0.3324 μg/mL(S/N=10)。
2.2.6 样品溶液精密度、稳定性试验 将2.2.2项混合对照品溶液进样,连续进样9次,计算得A~D杂质峰面积的RSD分别为0.29%、0.42%、0.73%、0.45%;每天进样,连续进样6 d,计算得A~D杂质峰面积的RSD分别为0.83%、1.78%、2.31%、1.88%。并分别于0、2、4、8、12 h进样,计算得不同时间点时A~D杂质峰面积的RSD分别为0.60%、1.07%、1.89%、1.26%。表明在常温条件下,本方法日内、日间精密度良好,12 h内除虫脲原料药溶液稳定性良好。
2.2.7 有关物质检查 取三批除虫脲原料药,按照2.2.2项的方法配制供试品溶液浓度为500 μg/mL进样测定并记录峰面积。测定结果如表3所示,用外标法计算除虫脲主成分含量均大于99%,以主成分自身对照法计算有关杂质的量,原料药杂质峰面积的总和均低于0.1%的鉴定限。
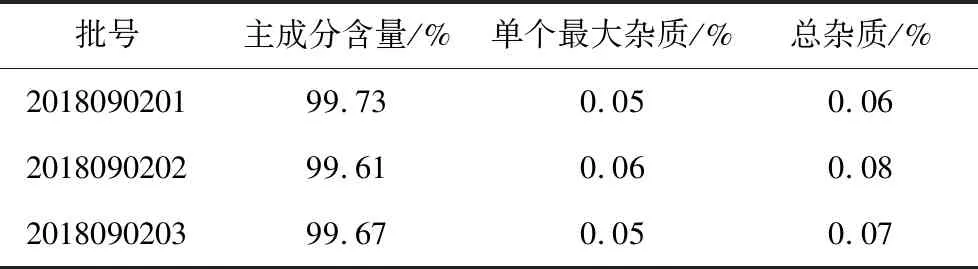
表3 除虫脲主成分含量和有关杂质
3 讨论与结论
3.1 衍生化方法的选择 根据文献[10-12]发现对氯苯基异氰酸酯在含有水相的溶液中不稳定,易水解,进样后在流动相中会发生水解影响检测,需要在进样前进行衍生化处理成在流动相中相对稳定的物质后,再进样检测。衍生化方法分别考察了两种常见的方法:一为对氯苯基异氰酸酯用四氢呋喃溶解后用过量二正丁胺进行衍生;二为用甲醇与对氯苯基异氰酸酯进行酯化反应的衍生方法。其中衍生化方法二的处理过程简单,易反应完全,且无溶剂峰和杂峰,不影响除虫脲及相关杂质的分析。因此,选择了甲醇与对氯苯基异氰酸酯进行酯化反应的衍生方法。
3.2 色谱条件的选择 本试验根据文献[13-15],流动相分别考察了乙腈-水-二恶烷、甲醇-水、甲醇-乙酸铵水这3种流动相体系。结果显示在乙腈-水-二恶烷的流动相条件下,对氯苯基异氰酸酯的峰型不佳,有拖尾且各峰分离不佳;在甲醇-水的流动相条件下,2,6-二氟苯甲酰胺的峰型不佳,有拖尾且各峰分离一般;甲醇-乙酸铵水的流动相条件下,除虫脲与2,6-二氟苯甲酰胺、对氯苯基异氰酸酯、对氯苯胺及1,3-双(4-氯苯基)尿素的峰型良好且各峰分离良好。因此,选择甲醇-乙酸铵水作为流动相体系。试验发现将流动相等度洗脱调整为梯度洗脱后,既能短时高效的检测出兽用除虫脲原料药是否引入其合成工艺上的杂质,同时能测定出主成分含量和有关物质,且各峰分离度良好。试验发现检测波长调整为254 nm时,各峰响应值均较高。
3.3 PDA检测色谱图分析 通过破坏性试验发现了除虫脲的多种有关物质,将降解产物R2和对氯苯胺的PDA检测色谱图进行对比分析,两张图谱最佳波长几乎一致,光谱学上说明强酸、强碱、高温和自然光破坏条件下的降解物R2为对氯苯胺,若完全确定,可做进一步的结构确证研究,其生成途径也有待进一步的解析。
药物有关物质的检查与鉴定在其质量标准的制定实施中十分的重要,但目前除虫脲未被药典收录且无有关物质的控制标准。本研究对除虫脲含量和有关物质液相方法建立的同时,对其有关物质进行了初步的研究。本研究发现3批次的除虫脲原料药中总杂质最大限度为0.1%,均低于《兽用化学药物杂质研究技术指导原则》中兽医专用原料药的杂质报告限度0.1%,鉴定限度0.2%,质控限度0.5%的标准。因此,依据现行传统工艺水平可暂定为如有杂质峰,各杂质峰面积的面积和不得大于对照溶液主峰面积的0.1倍(0.1%)。本研究建立测定除虫脲原料药主成分含量及有关杂质的高效液相色谱法,使除虫脲及有关杂质在同一色谱条件下检测并取得良好的分离效果,PDA检测图谱为除虫脲有关物质的定性分析提供了参考依据。该方法含量测定准确灵敏,杂质分离度好,结果准确可靠,可作兽用除虫脲原料药主成分含量及有关物质的检测与分析,为兽用除虫脲原料药的质量控制和质量标准制定提供了参考依据。
