加权ceRNA网络筛选乳腺癌生物标志物
2020-04-15朱东月
朱东月,朱 平
(江南大学理学学院,中国江苏 无锡 214122)
乳腺癌是全球女性发病率最高的癌症之一,相关数据显示大约每18 秒就有一名女性被确诊为乳腺癌[1]。乳腺癌是女性死亡的主要原因之一,占全球癌症死亡人数的14%[2]。目前,对于乳腺癌的诊断仍缺乏高效敏感的分子标志物[3]。因此,探索相关生物标志物对乳腺癌的诊断尤为重要。
癌症的大多数生物标志物主要是基于高通量基因组学数据分析获得的。RNA 测序技术的发展有助于揭示基因组的复杂性。已有资料显示,超过90%的基因组可以被转录,但只有1%~2%的转录产物可以编码蛋白质,而大多数转录产物是长链非编码RNA(long noncoding RNA,lncRNA)和微 RNA (microRNA,miRNA)[4]。lncRNA 是一类重要的非编码RNA,长度约为200 个核苷酸,由RNA 聚合酶Ⅱ转录,经选择性剪切加工而成熟。lncRNA 在复杂的生命过程中扮演着重要的调控角色,如参与调节免疫反应、恶性细胞的增殖和转移[5]。为了揭示 lncRNA 的功能,Zhang 等[6]通过构建lncRNA-mRNA 网络,发现了7 个与心肌梗死发生和复发相关的lncRNA。miRNA 是一类重要的非编码小RNA,其长度约为22 个核苷酸,可以通过与碱基配对来指导复合物识别mRNA 的3'端非翻译区,从而对靶基因起到调控的作用[7]。竞争性内源RNA(competing endogenous RNA,ce-RNA)假说主要是指不同类型的RNA 之间通过竞争一个或多个miRNA 的结合位点来调控基因表达[8]。ceRNA 在很多癌症的预后和治疗中都发挥着重要的作用。在胃癌中,lncRNA HOTAIR 通过结合 miR-331-3P 抑制 HER2 的表达,且 HOTAIR/HER2 的表达水平与晚期胃癌具有显著相关性[9];在肾透明细胞癌中,PTENP1 作为竞争性内源RNA 抑制癌症的发展[10];在乳腺癌中,linc-ROR通过调节miRNA 促进癌细胞的发展和转移[11]。
本文利用差异 mRNA、lncRNA 和 miRNA 构建ceRNA 网络,并结合网络的拓扑性质和RNA的表达数据计算该网络节点的权重,然后基于随机森林的改进算法筛选出一组RNA,该组RNA在识别乳腺癌方面具有较高精度和效率,结合其生物功能分析发现,该组RNA 可作为乳腺癌的生物标志物。
1 材料与方法
1.1 RNA差异表达分析
TCGA 数据库[12](https://cancergenome.nih.gov/)提供了各种基因组的高通量数据,包括mRNA、lncRNA 和miRNA 的表达数据。首先,从TCGA数据库中下载与乳腺癌有关的mRNA、lncRNA 和miRNA 的表达矩阵。其中mRNA、lncRNA 表达数据包括113 个正常样本和1 109 个乳腺癌样本,miRNA 表达数据包含76 个正常样本和752 个乳腺癌样本。然后,利用R 软件中的edgeR 包[13]对RNA 数据进行标准化和差异分析,以获得差异mRNA、lncRNA 和miRNA。在筛选差异表达mRNA 时,将筛选参数设置为:|log2FC|>2,P<0.001; 在筛选差异lncRNA 和差异miRNA 时,将筛选参数设置为:|log2FC|>1.5,P<0.05。
1.2 ceRNA网络的建立
本文研究的ceRNA 网络包含lncRNA-miRNA和miRNA-mRNA 两种靶向关系。miRcode 数据库[14](http://www.mircode.org/)覆盖了完整的GENECODE 注释的转录组,可用于预测lncRNA 和miRNA 之间的靶向关系。通过该数据库,获得了本研究所需的lncRNA-miRNA 关系对。此外,使用 miRDB(http://mirdb.org)、miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/)和 TargetScan (http://www.targetscan.org/vert_72/)在线分析工具[15~17]预测mi-RNA 的靶基因mRNA。为了提高生物信息分析的可靠性,本文利用3 个数据库中重叠的miRNA-mRNA 关系对进行后续的生物学分析。基于获得的 lncRNA-miRNA 和 miRNA-mRNA 关系对,利用Cytoscape 软件[18]将ceRNA 网络可视化。
1.3 ceRNA网络节点权重的计算
边聚集系数(edge clustering coefficient,ECC)是由Watts 等[19]于1998年提出的,用于衡量网络中一个节点与其邻接节点之间的亲疏程度,是描述网络拓扑性质的重要特征之一。根据Watts 等[19]对于边聚集系数的定义,ceRNA 网络的边聚集系数E(x,y)可以用下式表示:
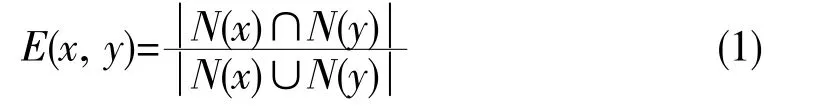
式中N(x)表示节点x 邻接节点的集合,N(y)表示节点y 邻接节点的集合。由上式可以看出,E(x,y)是一个局部指标,用于衡量网络中x 和y 两个节点之间的亲疏程度。E(x,y)的取值范围为[0,1],其值越大表示两个节点之间的关系更为紧密,属于同一模块的可能性越大。
皮尔逊相关系数(Pearson correlation coefficient,PCC)可用于筛选ceRNA 网络中的mRNA-lncRNA关系对[6]。该系数可用如下公式表示:
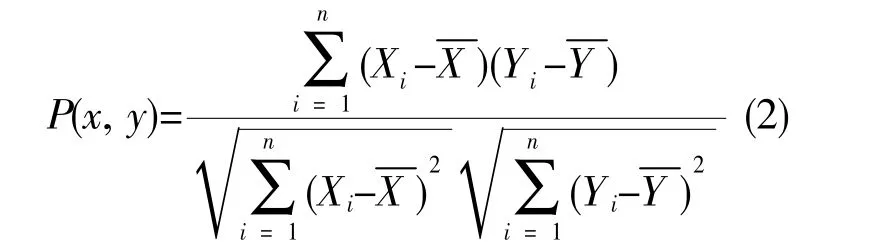
其中n 表示乳腺癌样本数,Xi表示在i 样本中x的表达量,Yi表示在i 样本中y 的表达量,分别表示在样本中x 和y 的平均表达量。使用值描述x 和y 相关性的强弱。的取值范围为[0,1],该值越大,mRNA 和lncRNA 的相关性越强。
在ceRNA 网络中,ECC 值和PCC 值较高的节点对所在模块的调控能力较强,该节点更倾向成为生物标志物[20]。因此,当衡量一个节点在网络中的调控程度时,需要综合考虑ceRNA 网络的拓扑性质和基因的表达相关性。利用下式定义每个节点的权重值:
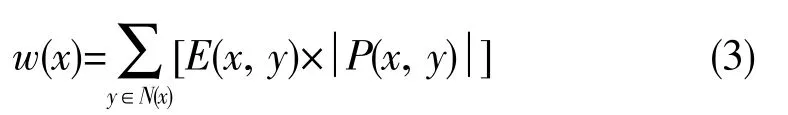
1.4 基于随机森林的逐步特征选择方法
随机森林(random forest,RF)已经广泛用于癌症生物标志物的分类和预测[21~22]。该算法可以避免过拟合且不受数据维度的影响,具有较好的分类精度和预测效果,但不可以降低变量子集的维度。本文将RF 改进为基于随机森林的逐步特征选择(stepwise feature selection based on random forest,SFS-RF)算法,用于筛选高效率和低维度的乳腺癌生物标志物,具体步骤为:1) 运用具有5-折交叉验证的随机森林算法计算单个变量的分类精度,并按照精度由高到低进行排序; 2) 将每个变量逐次放入到变量集中,如果增加一个变量可以使得分类精度提高,则保留该变量; 反之,则终止计算。算法运行结束时,即可筛选出分类精度最高且数目最小的变量集,以及相应的分类精度。
将本文采用的SFS-RF 方法与LASSO (least absolute shrinkage and selection operator)[23]、主成分分析(principal component analysis,PCA)[24]进行比较,并利用准确率(accuracy,ACC)、灵敏度(sensitivity,SN)、特异性(specificity,SP)和马修斯系数(Matthews correlation coefficient,MCC) 4 个评价指标衡量变量集的分类能力。ACC、SN、SP 和MCC的计算公式如下所示:
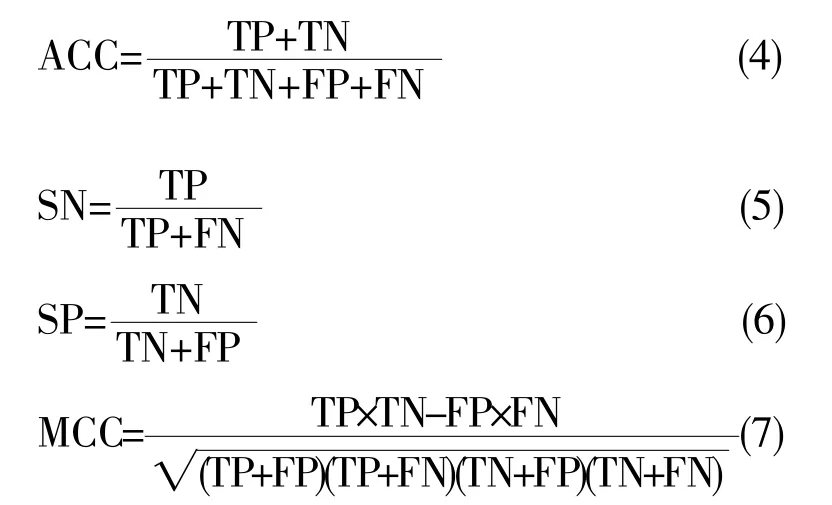
式中TP 表示真阳性,即预测为正样本实际也是正样本的数量; TN 表示真阴性,即预测为负样本实际也是负样本的数量; FP 表示假阳性,即预测为正样本实际为负样本的数量; FN 表示假阴性,即预测为负样本实际为正样本的数量。
同时,用受试者操作特征曲线(receiver operator characteristic curve,ROC 曲线)评价变量集的分类精度[25]。曲线下的面积(area under curve,AUC)用于数值化地反映算法的分类能力[25],其取值范围在[0,1],当AUC 值越接近1 时,模型的预测准确率越高。
1.5 富集分析
利用BiNGO 插件[26]对生物标志物调控的RNA进行GO (gene ontology)功能富集分析。GO 功能富集主要是从分子功能(molecular function,MF)、生物过程(biological process,BP)和细胞组分(cellular component,CC) 3 个层面探究RNA 的生物功能。利用 KOBAS 数据库[27](http://kobas.cbi.pku.edu.cn/)对生物标志物调控的RNA 进行KEGG (kyoto encyclopedia of genes and genomes)通路富集分析。在后续的分析中,P<0.05 的生物功能或者通路被认为具有统计学意义。
2 结果
2.1 差异表达的RNA
利用edgeR 包在正常样本和癌症样本中筛选出的差异表达mRNA、lncRNA 和miRNA 分别有847 个、438 个、152 个。其中,上调 mRNA 有 532个、下调 mRNA 有 315 个;上调 lncRNA 有 341个、下调lncRNA 有97 个; 上调miRNA 和下调 miRNA 依次有 128 个、24 个。
2.2 乳腺癌ceRNA网络
从miRcode 在线数据库中获得了99 个差异lncRNA-miRNA 相互作用关系对,其中包含27个差异 lncRNA 和 21 个差异 miRNA。利用miRDB、miRTarBase 和 TargetScan 数据库对差异miRNA 的靶基因mRNA 进行预测,获得差异miRNA-mRNA 相互作用关系对39 个,其中包括20 个差异miRNA 和 27 个差异mRNA。最终,通过lncRNA-miRNA-mRNA 关系对建立ceRNA 网络,该网络由 24 个 lncRNA、20 个 miRNA 和 27个 mRNA 组成(图 1)。在 ceRNA 网络中,节点表示差异mRNA、lncRNA 或者miRNA,边表示它们之间的联系。
2.3 SFS-RF方法筛选的生物标志物
权重不仅考虑了不同RNA 在ceRNA 网络中的邻接节点聚集情况,同时也考虑了RNA 之间表达的相关性。通过公式(3)计算ceRNA 网络节点的权重,发现的差异RNA 有27 个。用SFS-RF 方法对这27 个RNA 进行筛选,获得乳腺癌生物标志物,以用于高精度识别乳腺癌样本。最终从27个差异RNA 中筛选出一组生物标志物CHL1-AS2、LINC00466 和 LINC00337。
以这3 个RNA 作为识别乳腺癌样本的变量时,评价指标 ACC、SN、SP、MCC 和 AUC 的值依次为 0.98、0.85、0.98、0.84 和 0.98 (ROC 曲线见图2A),说明该组RNA 对识别乳腺癌样本具有很高的灵敏度和特异性。lncRNA、miRNA 和mRNA 均由基因转录而来,因此进一步利用GEO 数据库[28]验证该组RNA 对乳腺癌样本的识别情况。从GEO 数据库中下载基因芯片GSE75333 (包含3个正常样本和3 个乳腺癌样本),并用ROC 曲线检验这3 个差异lncRNA 识别乳腺癌的灵敏度和特异性。结果显示:CHL1-AS2、LINC00466 和 LINC00337 具有较高的识别精度(AUC=0.90)。此外,为了探究该组生物标志物在正常样本和乳腺癌样本中的表达情况,利用箱线图将它们在两组样本中的表达水平进行可视化。结果如图2B~D 所示,CHL1-AS2 在正常样本中显著高表达(P=6.8E-07);而LINC00466 和LINC00337 在正常样本中低表达,在癌症样本中显著高表达(P 依次为4.3E-59、7.4E-63)。这说明该组RNA 的异常表达可能与乳腺癌的发病机制相关。
2.4 方法比较
肿瘤基因表达具有多变量、高维度和高冗余的特点,为了获得有效且可靠的信息,LASSO 回归和PCA 已广泛运用于生物标志物的筛选[29~30]。根据TCGA 数据库中下载的乳腺癌RNA 数据,利用SFS-RF、LASSO 和PCA 方法筛选生物标志物,用 ACC、SN、SP、MCC 和 ROC 曲线衡量它们筛选生物标志物的分类精度。3 种方法筛选出的生物标志物数量及分类精度见表1。表1 直观地显示:在分类精度相近的情况下,SFS-RF、LASSO 和PCA 方法筛选出来的生物标志物数量依次为3、19 和9,显然SFS-RF 方法筛选出的生物标志物最少。在乳腺癌的研究中,通过本文提出的方法,只需检测3 个基因的转录水平就能达到与其他方法(如LASSO 回归和PCA)相近的识别精度。因此,SFS-RF 方法在筛选生物标志物时具有更高的效率。
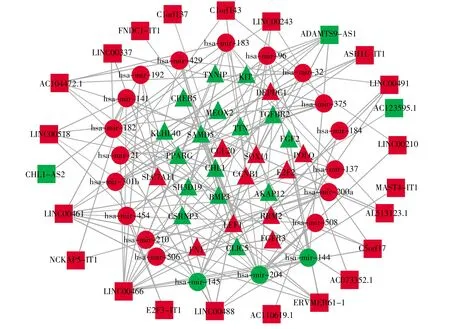
图1 乳腺癌ceRNA 网络正方形表示lncRNA; 圆表示miRNA; 三角形表示mRNA; 绿色节点表示下调RNA; 红色节点表示上调RNA。Fig.1 Breast cancer ceRNA networkSquares represent lncRNA; Circles represent miRNA; Triangles represent mRNA; Green nodes indicate down-regulated RNAs;Red nodes indicate up-regulated RNAs.
2.5 生物功能分析
在乳腺癌ceRNA 网络中,LINC00466、CHL1-AS2 和LINC00337 通过竞争性结合15 个miRNA调控32 个差异RNA 在乳腺癌中的表达(图3)。为了探究这些差异RNA 对乳腺癌发病机制的影响,对这些RNA 进行GO 和KEGG 富集分析。GO 分析结果显示,这些RNA 显著富集在染色体的形成、细胞增殖的正调控、蛋白激酶活性、姐妹染色单体的分离、调节磷酸盐代谢过程等387 个功能类别,其中在生物过程(BP)和分子功能(MF)中排名前10 的条目如图4A 所示。KEGG 通路分析结果表明,它们主要富集在TNF 信号通路、PI3KAkt 信号通路、MAPK 信号通路、细胞衰老和乳腺癌等15 条通路,排名前10 的富集通路见图4B。从上述结果可以看出,该组生物标志物通过调控ceRNA 网络中RNA 的表达,参与乳腺肿瘤细胞的增殖、遗传物质的合成和信号传导等生物过程。
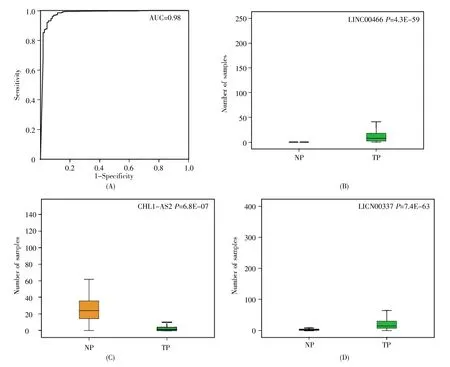
图2 生物标志物的ROC 曲线和差异表达(A) ROC 曲线; (B~D) 生物标志物在正常样本和乳腺癌样本中的差异表达。NP:正常样本; TP:癌症样本。Fig.2 ROC curve and differential expression of biomarkers(A)ROC curve;(B~D)Differential expression of biomarkers in normal and cancer samples.NP:Normal samples;TP:Cancer samples.
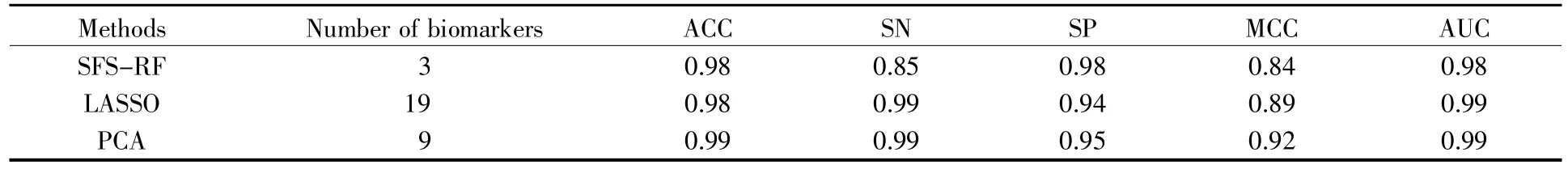
表1 SFS-RF、LASSO 和 PCA 方法的比较Table 1 Comparison of SFS-RF,LASSO and PCA methods
值得注意的是,图 3 显示 hsa-mir-145 与hsa-mir-183 通过一个差异lncRNA C5orf17 发生间接相互作用。由此,本文获得了一个有意义的miRNA—lncRNA—miRNA 调控轴,即 hsa-mir-183—C5orf17—hsa-mir-145。该调控轴是生物标志物 LINC00466、CHL1-AS2 和 LINC00337 相互调控的中间渠道。
3 讨论
乳腺癌是女性中发病率最高的恶性肿瘤之一,通过构建其加权ceRNA 网络,可以挖掘出与乳腺癌发生发展较为紧密的RNA,并且深入探讨这些RNA 参与的生物过程及其具有的生物功能,可以为乳腺癌的发病机制探究提供一定的理论支撑。本研究通过对乳腺癌的ceRNA 网络加权提取了27 个差异RNA,然后采用SFS-RF 方法筛选出一组可以作为乳腺癌生物标志物的RNA,即LINC00466、CHL1-AS2 和 LINC00337。GEO 数据集验证结果显示,这3 个差异lncRNA 在识别乳腺癌样本方面具有很高的可靠性和准确性(AUC=0.90)。
在这组RNA 中,LINC00466 已被证实可以作为乳腺癌的竞争性内源RNA,并参与乳腺肿瘤的发生、发展、增殖和转移等过程[31]。CHL1-AS2 和LINC00337 是本文发现的两个新的乳腺癌竞争性内源RNA。目前,暂未发现这两种RNA 在乳腺癌识别中的应用价值,但已有研究表明它们对子宫内膜异位症[32]、肺腺癌[33]和胃癌[34]的发生有一定的影响。Zhang 等[32]使用定量聚合酶链反应(qPCR)在异位子宫内膜中发现CHL1-AS2 的表达水平明显高于正常子宫内膜,故认为这可能与子宫内膜异位症的发生有关。LINC00337 位于1 号染色体,由5 个外显子组成,在肺腺癌ceRNA 网络中,LINC-00337 通过竞争性结合hsa-mir-373 和hsa-mir-519 调控PBK 和KIF23 的表达,进而调节免疫系统,最终影响患者的预后[33]。Hu 等[34]发现胃癌样本中LINC00337 的表达水平明显高于正常样本,并且其可利用BZH2 作为媒介抑制p21 表达,以促进胃癌细胞的增殖。因此,CHL1-AS2 和LINC00337 可能是乳腺癌潜在的生物标志物,值得进一步研究。
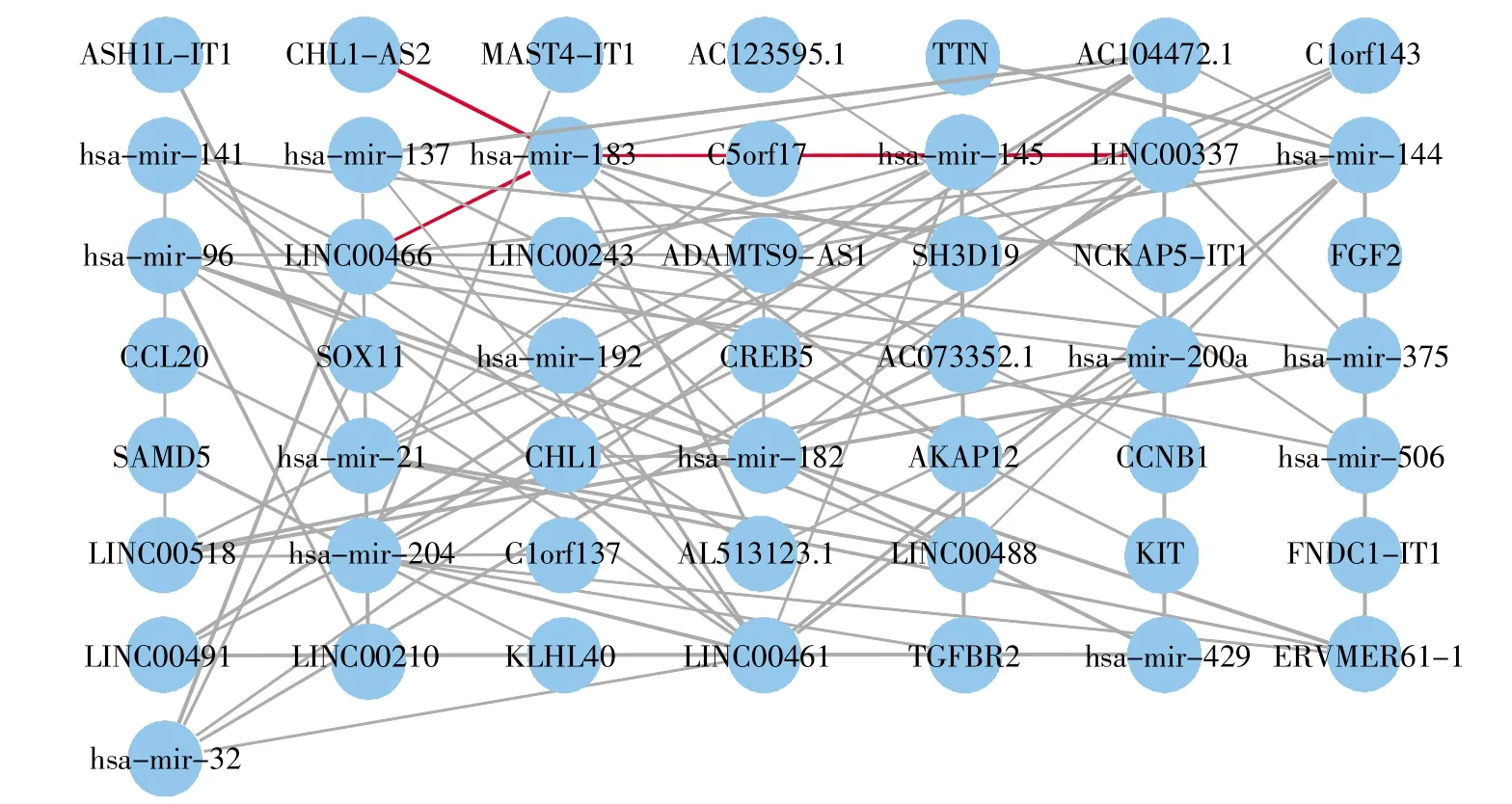
图3 生物标志物的ceRNA 子网络Fig.3 ceRNA subnetwork of three biomarkers
在GO 富集分析结果中,我们发现了几类与肿瘤细胞增殖、遗传物质合成及蛋白质代谢密切相关的功能注释,如浓缩染色体、细胞增殖的正调控、蛋白激酶活性和正调控高分子代谢等功能条目,说明 LINC00466、CHL1-AS2 和 LINC00337 通过竞争性地结合miRNA 调控多个RNA 参与细胞增殖、分化和遗传物质的合成等生物过程。在KEGG富集分析中,多条通路已被证实与乳腺癌的发生相关。肿瘤坏死因子(tumor necrosis factor,TNF)可以直接杀伤肿瘤细胞,相关研究表明当TRADD 低表达时,TNF 信号通路杀伤肿瘤细胞的作用减弱[35]。MALAT1 低表达可激活PI3K-Akt 信号通路,进而调节乳腺肿瘤细胞的转移[36]。MAPK 信号通路对乳腺癌的发生发展有着重要的影响,研究显示:EGFR 过表达激活MAPK 信号转导通路,活化原癌基因c-fos,刺激细胞恶性增殖[37]。KEGG通路富集结果与GO 富集结果相互验证,说明在生物功能和生物通路中,研究这些生物标志物是有意义的。同时,本文获得了一个有意义的调控轴 hsa-mir-183—C5orf17—hsa-mir-145,其可作为 LINC00466、CHL1-AS2 和 LINC00337 相互调控的中间渠道,且hsa-mir-183[38]和hsa-mir-145[39]已被证实与乳腺癌的发生、发展和预后等方面有着密切的联系。这些表明LINC00466、CHL1-AS2和LINC00337 可以作为生物标志物,用于高精度识别乳腺癌样本。
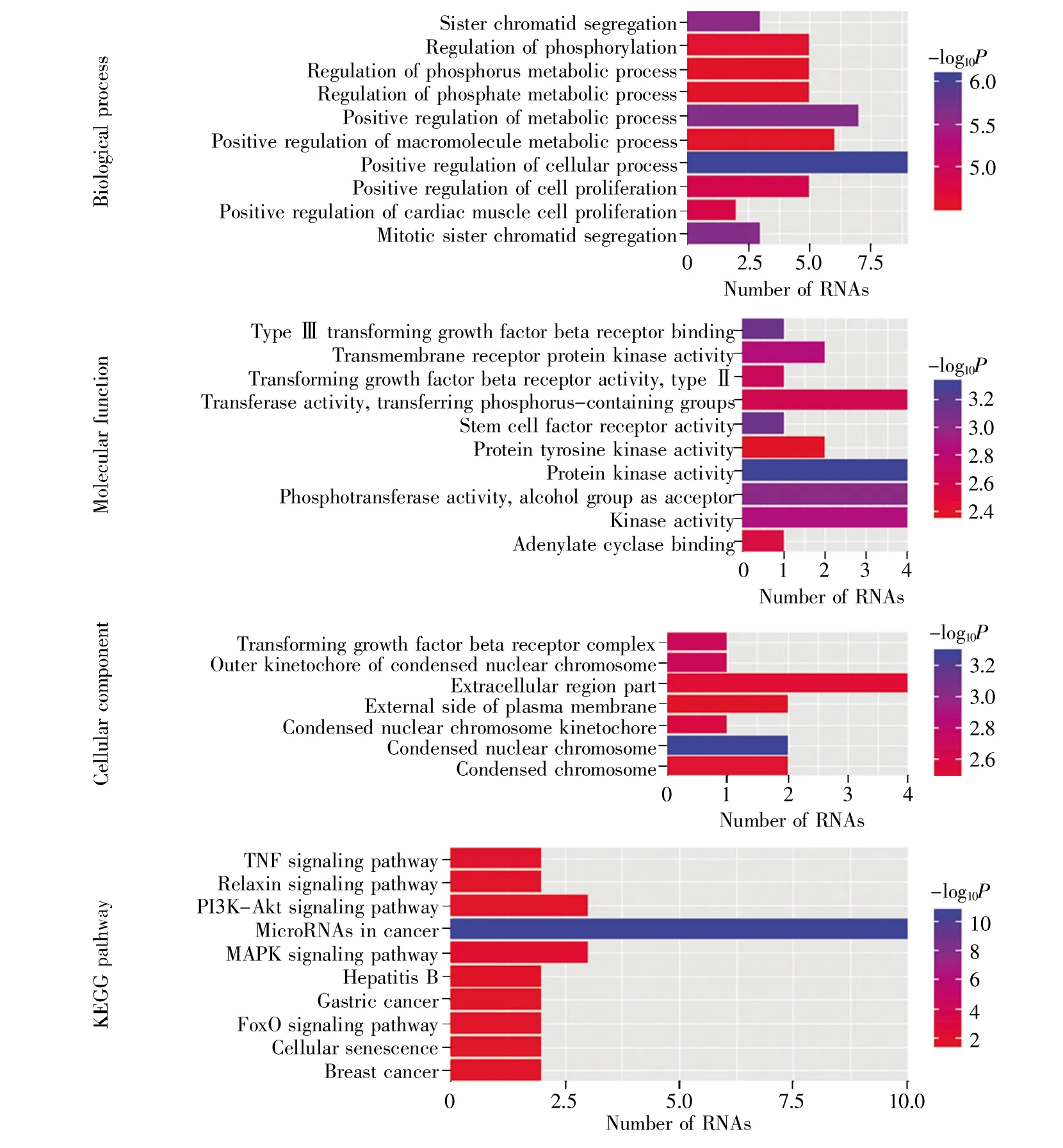
图4 GO 和KEGG 富集分析Fig.4 GO and KEGG enrichment analyses
总的来讲,本文构建了乳腺癌加权ceRNA 网络,并将其用于研究RNA 分子与乳腺癌发病机制之间的联系。其次,利用SFS-RF 方法,筛选出了一组可用作乳腺癌生物标志物的RNA——LINC00466、CHL1-AS2 和 LINC00337。其中,CHL1-AS2 和LINC00337 作为首次发现的标志物,为探究乳腺癌ceRNA 调控机制提供了新的思路。
