通过型固相萃取-超高效液相色谱-串联质谱法检测火锅底料中的喹诺酮类药物
2020-04-13沈雄雅何晓明吴丹虹叶冰洁闻佳钰刘强欣
沈雄雅 何晓明 吴丹虹 叶冰洁 闻佳钰 刘强欣


摘要 [目的]建立通过型固相萃取-超高效液相色谱-串联质谱法测定火锅底料中11种喹诺酮类药物的分析方法。[方法]样品用酸化乙腈提取,经Oasis Prime HLB固相萃取柱净化,收集流出液,用超高效液相色谱-串联质谱仪多反应监测扫描模式检测和同位素内标定量。[结果]所有目标物在线性范围内线性良好,相关系数≥0.99,检出限为0.6~4.8 μg/kg,定量限为2.0~13.2 μg/kg,不同加标水平的平均回收率为78.5%~103.4%,相对标准偏差为2.8%~9.1%。[结论]该方法具有快速、准确、灵敏度高的特点,可用于火锅底料中11种喹诺酮类药物的测定。
关键词 通过型固相萃取-超高效液相色谱-串联质谱,火锅底料,喹诺酮类药物
中图分类号 TS201.6文献标识码 A
文章编号 0517-6611(2020)06-0184-04
Abstract [Objective]The research aimed to establish an analytical method for the determination of 11 quinolones in hotpot seasoning by solid phase extractionultra performance liquid chromatographytandem mass spectrometry.[Method]Samples were extracted with acidic acetonitrile,cleanedup by soludphase extraction using Oasus Prime HLB column. The analytes were detected in the multireaction monitoring (MRM) mode and quantified by the isotope internal standard method. [Result]The calibration curves were linear well in the corresponding concentration ranges,with correlation coefficient ≥ 0.99. The limits of detection and quantification were 0.6-4.8 μg/kg and 2.0-13.2 μg/kg,respectively.The average recoveries at three spiked levels ranged from 78.5% to 103.4% with RSD of 2.8%-9.1%.[Conclusion]This method is rapid,accurate and sensitive.It can be used for the determination of 11 quinolones in hotpot seasoning.
Key words Solid phase extractionultra performance liquid chromatographytandem mass spectrometry,Hotpot seasoning,Quinolones
喹諾酮类药物是一类人工合成的广谱抑菌剂,具有抗菌谱广、高效、给药方便、价格低廉等特点,在临床治疗感染方面有较广泛应用[1-2]。有研究表明,消费者长期食用含有喹诺酮类药物残留的食物,会诱发致病菌产生耐药性,甚至还有潜在的致癌作用[3-4]。近年来,少数不法分子将喹诺酮类药物添加在火锅底料中用于杀菌,防止人们食用变质的该类食物后出现肠胃不适等情况,给食品安全及消费者健康带了巨大的隐患。因此,建立一种灵敏、准确、快速同时测定火锅底料中喹诺酮类药物的检测方法,对保障食品质量安全、进而保护消费者健康具有重要意义。
目前对喹诺酮类药物检测方法多集中在动物源性食品和环境等领域[5-9],关于火锅底料中喹诺酮类药物相关研究报道较少[10-12],净化方式主要采用液液萃取法、分散固相萃取法或固相萃取法等。液液萃取法、分散固相萃取法虽然简单,但基质效应大,固相萃取的Oasis HLB柱需要进行活化、淋洗、洗脱等处理,过程繁琐,消耗时间长。该试验采用通过型固相萃取技术净化样品,对前处理提取条件、净化条件和仪器分析条件进行了优化,采用超高效液相色谱-串联质谱检测,同位素内标定量,建立火锅底料中11种喹诺酮类药物的检测方法。
1 材料与方法
1.1 仪器 LC-MS/MS 8050三重四级杆液相色谱-串联质谱仪(日本Shimadzu公司), ST16R高速冷冻离心机(美国Thermo公司), Multi Reax全能型振荡器(德国Heidolph公司),AUTO EVA-60全自动平行浓缩仪(睿科公司), Milli-Q Reference超纯水机(美国Millipore公司)。
1.2 试剂 喹诺酮类标准物质:诺氟沙星、氧氟沙星、洛美沙星、培氟沙星、氟罗沙星、沙拉沙星、双氟沙星、司帕沙星、环丙沙星、达氟沙星、恩诺沙星(纯度均大于95%,德国 Dr.Ehrenstorfer 公司),同位素内标:恩诺沙星-D5、环丙沙星-D8、诺氟沙星-D5(纯度均大于99%,德国Witega公司)。
甲酸、乙腈(HPLC级,美国Thermo公司),Oasis Prime HLB固相萃取柱(60 mg/3 mL,美国Waters公司),甲酸铵(天津博纳艾杰尔科技有限公司),试验用水为Milli-Q超纯水,其他试剂均为国产分析纯试剂。
1.3 标准溶液的配制 分别将11种喹诺酮类药物标准品配制成浓度为100 mg/L的单标准储备溶液,于4 ℃ 下避光保存。取各单标准储备液,用甲醇稀释成10 mg/L的混合标准中间液,用时根据需要,将混合标准中间液用初始浓度流动相逐级稀释配成系列混合标准溶液,现配现用。
0.1 mol/L Na2EDTA-Mcllvaine溶液:准确量取1 000 mL 0.1 mol/L 柠檬酸溶液与 625 mL 0.2 mol/L 磷酸氢二钠溶液混匀,用NaOH 调至 pH 4后,再加入 60.5 g 乙二胺四乙酸钠,摇匀。
1.4 样品前处理
1.4.1 提取。
准确称取5.0 g的样品于50 mL离心管中,加入20 mL 0.1 mol/L Na2EDTA-Mcllvaine 缓冲溶液,涡旋混匀1 min后,加入10 mL 1%甲酸乙腈,涡旋振荡15 min,10 000 r/min高速离心3 min。 取上清液于15 mL离心管中,待净化。
1.4.2 净化。准确移取2 mL样品提取液,加于Oasis Prime HLB固相萃取柱上,控制流速为1滴/s,收集全部流出液,40 ℃ 氮吹至干,用1 mL初始浓度流动相复溶,经0.22 μm滤膜过滤后,待上机测定。
1.5 测定方法
1.5.1 色谱条件。
色谱柱:ACQUITY UPLC HSS T3(2.1 mm×100 mm,1.8 μm),柱温40 ℃,流速0.35 mL/min,进样量3 μL,流动相:A相为2 mmol/L甲酸铵水溶液(含0.1%甲酸),B相为乙腈,梯度顺序:0~0.5 min,7% B,0.5~3.0 min,7%~25% B,3.0~5.0 min,25%~95% B,5.0~5.9 min,95% B,5.9~6.5 min,95%~7% B。
1.5.2 質谱条件。
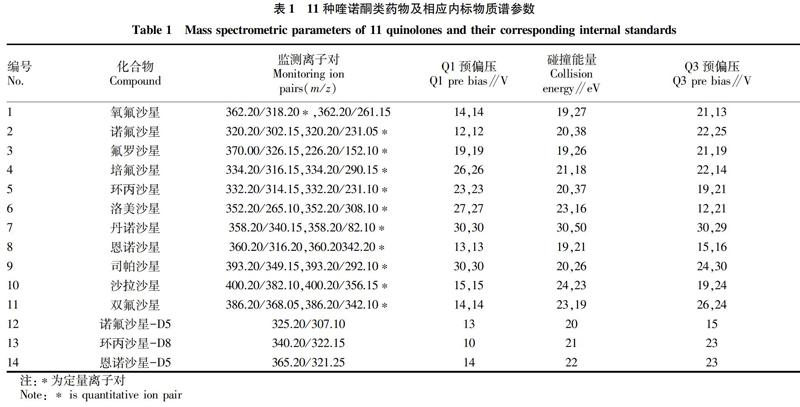
电喷雾离子源(ESI):正离子模式,温度400 ℃,毛细管电压 4 000 V,雾化气流量3.0 L/min,加热气流量10.0 L/min,定性与定量离子对、碰撞能量等参数见表1。
2 结果与分析
2.1 分析条件的优化
在电喷雾正离子模式下,采用直接进样方式对11种喹诺酮类药物进行全扫描,确定分子离子峰。再以分子离子峰作为母离子,进行子离子扫描,确定每个目标化合物的定性和定量离子对,最后通过多级反应监测模式,对各化合物定性定量离子对及碰撞能等参数进行优化。
在流动相中添加缓冲盐或甲酸是改善色谱峰形、提高仪器响应值和离子化效率的常用有效手段[13]。该试验比较了甲醇-0.1%甲酸水溶液、甲醇-2 mmol/L甲酸铵水溶液(含0.1%甲酸)、乙腈-0.1%甲酸水溶液和乙腈-2 mmol/L甲酸铵水溶液(含0.1%甲酸)的分离效果。结果表明,甲醇和乙腈对目标化合物响应值无显著差异,但甲醇的黏度大,作为流动相时色谱柱柱压较高,保留时间延长。加入2 mmol/L甲酸铵后,色谱峰峰型更尖锐,对称性更好。选择乙腈-2 mmol/L甲酸铵水溶液(含0.1%甲酸)作为流动相时获得的11种喹诺酮类药物的色谱图见图1。
2.2 提取和净化条件优化
2.2.1 提取条件的优化。
喹诺酮类药物的分子结构中均含有羧基和叔氨基[14],易溶于酸性或碱性溶液中,通常采用酸性乙腈溶液或酸性甲醇溶液作为提取溶剂。考虑到火锅底料中含有较多的油脂和蛋白质[15],而乙腈具有良好沉淀蛋白的作用,带出的极性成分较少,能在一定程度上减少基质效应。
火锅底料样品黏度大,在乙腈提取前需要对样品进行分散,增加后续提取溶剂与样品的接触面积。该试验比较了超纯水和0.1 mol/L Na2EDTA-Mcllvaine 缓冲溶液对样品的回收率的影响。结果表明,0.1 mol/L Na2EDTA-Mcllvaine 缓冲溶液的提取效率明显好于超纯水,这是因为Na2EDTA作为是一种良好的螯合物,可与样品中的金属离子络合,使目标化合物游离出来,从而提高提取效率[16]。因此,该试验确定先用0.1 mol/L Na2EDTA-Mcllvaine 缓冲溶液分散样品,再以1%甲酸乙腈提取。
2.2.2 净化条件的优化。
火锅底料基质复杂,为提高检测灵敏度,降低基质干扰,需对提取液进行净化。该试验考察了液液萃取[11]、分散固相萃取[10]、固相萃取3种净化方式,并选择Oasis HLB [12]和Oasis Prime HLB等不同类型的固相萃取柱进行净化效果的比较。结果表明(图2),液液萃取法和分散固相萃取法虽然方法简单,但净化效果不理想,净化液较混浊,回收率偏低,使用Oasis HLB及 Oasis Prime HLB固相萃取柱净化后,净化液澄清透明,回收率均大于80%,但Oasis HLB固相萃取柱需经活化、上样、淋洗和洗脱等多个步骤,操作繁琐,而Oasis Prime HLB固相萃取柱可以一步法净化,在保证净化效果的同时简化了前处理步骤,提高检测效率。因此,净化方法选择Oasis Prime HLB固相萃取柱净化。
2.3 基质效应 样品中基质成分会对分析过程有显著的干扰,并影响分析结果的准确性,这些影响和干扰被称为基质效应[16]。该试验采用提取后添加法,测定空白基质提取液与纯溶剂中同浓度目标化合物的离子响应强度,通过二者比值来评价基质效应(ME)。结果表明(表2),在经过优化样品前处理方法后,部分喹诺酮类药物仍然存在不同程度的基质抑制效应。该试验采用同位素内标法进行定量,一方面可以补偿基质效应,一方面也减少了样品处理过程中可能带来的误差,提高定量准确性。
2.4 线性范围、检出限和定量限 配制6 个浓度5、10、25、50、100和200 μg/L为混合标准工作溶液,内标物浓度均为25 μg/L,在优化的分析条件下,进行测定。以各目标化合物与相应内标物峰面积的比值(y)为纵坐标、质量浓度(x)为横坐标,绘制标准曲线,得到线性回归方程和相关系数。
对空白火锅底料样品进行低水平加标试验,计算信噪比(S/N)。以3倍信噪比和10倍信噪比对应的浓度为检出限(LOD)和定量限(LOQ)。11种喹诺酮类药物的线性方程、相关系数、LOD和LOQ见表2。
2.5 回收率与精密度
选取空白火锅底料样品进行了3个不同浓度水平的加标回收试验,6次平行试验的结果见3。从表3可看出,在添加浓度范围内,11种喹诺酮类药物的平均回收率为78.5%~103.4%,相对标准偏差(RSD)为2.8%~9.1%。结果表明该方法具有较好的回收率,可以满足日常检测要求。
2.6 实际样品的分析
应用该方法对15 个不同品牌和批次的火锅底料进行测定,结果发现11种喹诺酮类药物均未检出。
3 结论
该试验建立了通过型固相萃取-超高效液相色谱-串联质谱法测定火锅底料中11种喹诺酮类药物的分析方法,优化了色谱质谱条件以及提取净化条件。与传统固相萃取技术相比,该方法简便、快速、检测效率高,能很好地应用于日常大批量检测。
参考文献
[1] 岳振峰,林秀云,唐少冰,等.高效液相色谱-串联质谱法测定动物组织中的16种喹诺酮类药物残留[J].色谱,2007,25(4):491-495.
[2] 林黎,張毅,涂小珂,等.液相色谱-串联质谱法同时测定化妆品中的25种喹诺酮类药物[J].色谱,2015,33(3):275-281.
[3] 李婧妍,郭春锋,崔立辉,等.QuEChERS HPLC-MS/MS法测定禽肉中9种喹诺酮类兽药残留量[J].食品研究与开发,2016,37(22):153-157.
[4] 梁素丹,陈剑刚,张艳.全自动固相萃取-超高效液相色谱-串联质谱法同时测定动物肌肉中喹诺酮类和四环素类兽药残留[J].中国食品卫生杂志,2018,30(2):151-157.
[5] 刘柏林,谢继安,赵紫微,等.超高压液相色谱-电喷雾串联四极杆质谱内标法同时测定禽类食品中11种喹诺酮类药物[J]. 中国食品卫生杂志,2017,29(3):316-321.
[6] 尹燕敏,沈颖青,朱月芳,等.超高效液相色谱-串联质谱法同时测定水和沉积物中磺胺类、喹诺酮类和氯霉素类抗生素残留[J].分析科学学报,2015,31(2):228-232.
[7] CAO G Z,ZHAN J,SHI X Z,et al. Analysis of 140 veterinary drugs and other contaminants in poultry muscle by ultrahighperformance liquid chromatography-tandem mass spectrometry [J]. Chromatographia,2018,81 (4):707-718.
[8] HE Z Y,WANG Y H,XU Y P,et al. Determination of antibiotics in vegetables using QuEChERSbased method and liquid chromatography-quadrupole linear ion trap mass spectrometry [J]. Food analytical methods,2018,11(10):2857-2864.
[9] 陈磊,吴赟琦,赵志勇,等.QuEChERS/超高效液相色谱-串联质谱法快速测定土壤中19种氟喹诺酮类抗生素残留[J].分析测试学报,2019,38(2):194-200.
[10] 曹鹏,牟妍,高飞,等.分散固相萃取-超高效液相色谱-串联质谱法同时检测火锅食材中11种喹诺酮类药物[J].色谱,2013,31(9):862-868.
[11] 冯月超,王颖,王建凤,等.液质联用法同时测定火锅调料中15种喹诺酮类抗生素[J].分析试验室,2016,35(8):924-927.
[12] 国家市场监督管理总局.豆制品、火锅、麻辣烫等食品中喹诺酮类化合物的测定:BJS 201909[S].北京:国家市场监管总局,2019.
[13] 董茂锋,白冰,唐红霞,等.高效液相色谱串联质谱法测定玉米及植株中胺唑草酮及其代谢物[J].分析化学,2015,43(5):663-668.
[14] 刘芃岩,申杰,刘磊.复合模板印迹聚合物净化液相色谱-质谱联用法测定鱼肉中氟喹诺酮类残留[J].分析化学,2012,40(5):693-698.
[15] 李兴根,乔勇升,陈伟,等.超高效液相色谱-串联质谱法快速测定火锅底料中的5种罂粟壳生物碱残留[J].分析测试学报,2018,37(4):446-451.
[16] 林静,张顺,蔡挺,等. QuEChERS-超高效液相色谱-串联质谱技术同时测定大蒜中10种农药残留[J].浙江农业学报,2018,30(1):159-166.
